There are approximately 7,000 rare diseases that are determined by a single gene. It was only in the 1980s that investigation of the causes of such genetic diseases and the quest for finding therapies started. Since then the genetic origin of over 5,800 rare diseases has been identified. Understanding the function of individual genes involves chromosome typing, genetic sequencing, molecular methods and animal models, all of which are now considered routine. However, it was not the case ten years ago, when the Nephrogenetic Research Group at Semmelweis University’s 1st Department of Paediatrics led by Dr. Kálmán Tory set out to track down the origins of an unknown disease.
Locating the causal gene
 The starting point of the research were two cousins treated by Dr. Attila Szabó, Professor at Semmelweis University’s 1st Department of Paediatrics, who were suffering from an unknown disease affecting mainly the male members of the family for the past three generations. The patients had nephrotic syndrome with cataracts, hearing impairment and enterocolitis in addition to the stagnation of growth from the age of 6 months and death at a very young age.
The starting point of the research were two cousins treated by Dr. Attila Szabó, Professor at Semmelweis University’s 1st Department of Paediatrics, who were suffering from an unknown disease affecting mainly the male members of the family for the past three generations. The patients had nephrotic syndrome with cataracts, hearing impairment and enterocolitis in addition to the stagnation of growth from the age of 6 months and death at a very young age.
Female members of the family were also affected by the disease, however with milder symptoms; they had cataracts and hearing impairment but their condition was not life threatening. Such difference in the severity of the disease in males and females suggested that it was inherited in an X-linked recessive pattern.
By comparing the X chromosomes of healthy and affected family members (chromosome typing) in collaboration with the Institute Imagine Paris, the researchers managed to pinpoint the region of the X chromosome responsible for the disease. This region still held 120 genes that had to be analysed to locate the origin of the disease. The identification of the gene was sped up by an unexpected event: two male family members of the second generation were found who were carriers of the region but were healthy. This discovery suggested that the mutation in the region of the X chromosome appeared during the formation of the germs cells of the great-grandmother and provided a perfect basis for locating the gene carrying the variation.
“This family member, who was a healthy carrier, was the point of reference for DNA sequencing: the region of interest in his X chromosome and that of an affected family member were sequenced. The result confirmed our previous expectations: we found only one variant to be different, the causal one. The DNA samples were sequenced in Germany at the University of Cologne as we did not have the technology ten years ago. Today, both chromosome typing and DNA sequencing would be possible in Hungary as well.”, said Dr. Kálmán Tory about identifying the gene responsible for the disease.
Proving the origin of the disease by protein structure studies and an animal model
Upon locating the gene causing the disease, the researchers were struck by an unexpected discovery: the gene had already been described in a completely different disease, dyskeratosis congenita. The mutations of the very same gene caused abnormalities on the tongue, nails, skin and in the bone marrow and did not cause early death in patients. To reveal the unusual mechanism of the newly discovered disease, protein structure studies were performed by Dr. Dóra K. Menyhárd at the Protein Modelling Group of Eötvös Loránd University.
 “The studies analyzed the protein complex affected by the genetic variation and showed that the mutation causes changes in the interaction of dyskerin and NOP10 proteins. The protein dyskerin has two functions: modifying RNA and maintaining telomeres, the protective caps of the chromosomes, which get shorter by age. In case of the disease we identified, the region responsible for RNA modification was damaged, while dyskeratosis congenita is attributed to telomere attrition.”, said Dr. Kálmán Tory.
“The studies analyzed the protein complex affected by the genetic variation and showed that the mutation causes changes in the interaction of dyskerin and NOP10 proteins. The protein dyskerin has two functions: modifying RNA and maintaining telomeres, the protective caps of the chromosomes, which get shorter by age. In case of the disease we identified, the region responsible for RNA modification was damaged, while dyskeratosis congenita is attributed to telomere attrition.”, said Dr. Kálmán Tory.
Finding the gene causing the variation took only two years, however, functional studies lasted ten years due to the lack of suitable technology that would have sped up the process. To prove that the disease was caused by the dysfunction of dyskerin in modifying RNA, an animal model using zebrafish was created together with the research group of the Department of Genetics at Eötvös Loránd University led by Dr. Máté Varga. The equivalent of the respective human gene was knocked out in the fish and the resulting mutant fish could be used to trace back the changes related to the disease.
“We saw that the zebrafish developed a similar disease to that of the patients: it affected the eyes, ears and kidneys of the fish, they ceased to grow at a certain point and died early on. We examined the telomeres at the time of the death of the animal and found that they had not got shorter yet. Just like in the case of the patients’ cells: upon the death of the fish, the RNA damage was already present without telomere attrition. We believe, the affected boys would have shown symptoms of dyskeratosis congenita later on as well, but its symptoms appear at the end of the first decade of life and the patients died at a much younger age. With the animal model we confirmed the causal role of the variant and what kind of dysfunction of the damaged protein complex was responsible for the disease. ”, said Dr. Kálmán Tory about the results of the functional studies on animal models.
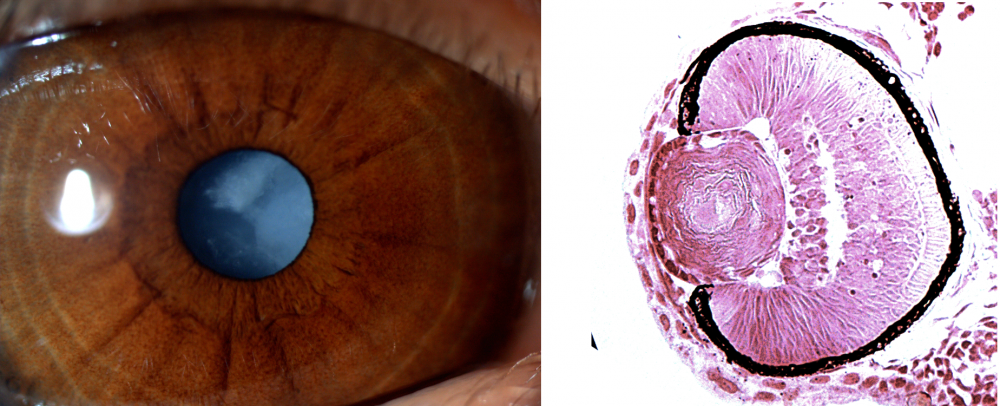
Finding another family affected by the disease
Another unexpected event gave a new twist to the research. Two years ago, a research group from the University College London contacted the Institute Imagine in Paris with a newly discovered nephrosis and were looking for other affected families. The Hungarian and English research groups made contact through the French institution and compared their findings realizing that they were researching the same disease. The English group found a mutation in NOP10 that damaged the binding of dyskerin and NOP10 proteins similarly to the variant identified by the Hungarian researchers.
Today, there are only two known cases of the disease: the Hungarian family who had been under treatment at Semmelweis University and the British family, who had been cared for at the University College London. Even though treatment is not available yet, affected families are now able to see and understand why the disease develops and how it is inherited. Knowing the genetic variant is also an important reference point in genetic consultations. The results have also proven that clinical observations may lead to extensive basic research, which can increase our knowledge of pathogenetic mechanisms. By finding the pathogen and unveiling the underlying mechanisms, this study is another step towards the better understanding of rare genetic diseases.
Ágnes Raubinek
Photos: Attila Kovács – Semmelweis University, MTA-SE Nephrogenetic Research Group