Szerkesztő: Guczogi Annamária
A világ legdrágább gyógyszere, egy kis magyar kórház, hétköznapi hősök, egy ország összefogása, határok nélküli gyógyítás… mozaikdarabkák egy olyan képből, amelynek végső formáját még senki nem ismeri.
A cikk elkészüléséhez Dr. Hantos Mónikával a MRE Bethesda Gyermekkórházának főgyógyszerésznőjével beszélgettem, és Dr. Magyar Bernadett gyógyszerész rezidens szakdolgozatát, valamint Tóth Krisztinának, Györök Zente édesanyjának fia fejlődéséről írt facebook oldalát használtam fel.
MOZDULATLAN TESTBE ZÁRVA…
2019 őszén még a koronavírus járvány kitörése előtt talán nem volt olyan ember, aki ne hallott volna Györök Zentéről, és a bűvös 700 millió forintról, amiért a szülei gyűjtést indítottak. Ekkor került reflektorfénybe maga a betegség is, az SMA. Az angol betűszó kifejtése spinalis muscularis atrophia, azaz gerincvelő eredetű izomsorvadás. Ez a ritka genetikai betegség a gerincvelői elülső alfa-motoneuronok pusztulásával jár, és minden 35. ember hordozza, Magyarországon évente 12-15 SMA-s gyermek születik. Oka a SMN1 gén 7. és 8. exonjának hiánya, vagy pontmutációja, ami miatt nem termelődik az SMN fehérje, ez pedig a motoneuronok elhalását okozza, és így végül beidegzés hiányában az izmok elsorvadnak. A folyamat kiterjed a szívre, májra, tüdőre, súlyos esetben végül légzéselégtelenség miatt a beteg halálához vezet.

A betegségnek 4 típusát különbözteti meg a szakirodalom, attól függően, hogy milyen korban jelennek meg a tünetek (izomhypotonia, mozgásszegénység, légzészavar…), és ezek milyen fokúak. SMA-1 esetében fél éves kor előtt, SMA-2 esetében fél éves kor után, SMA-3-nál 1,5 éves kor körül SMA-4-nél pedig csak felnőttkorban jelentkeznek a tünetek. SMA-1 betegek nem képesek önállóan ülni, és általában 2 éves kor előtt életüket vesztik, SMA-4 esetében csak enyhébb izomgyengeség, járászavar jelentkezik, amely az élettartamot nem befolyásolja. A betegség súlyosságát az SMN2 gén kópiaszáma határozza meg. Ez az úgynevezett „tartalék gén” képes valamennyi fehérjét termelni, tehát minél kevesebb van ebből, annál súlyosabbak a tünetek.
A betegség autoszomális recesszív módon öröklődik, így ha két hordozó (akik adott esetben nem is tudnak erről) családot alapít, akkor 50 % az esélye, hogy a gyermekük hordozó, 25 % hogy egészséges és 25 % hogy beteg lesz. A probléma sok esetben, hogy későn születik diagnózis, mivel a betegség nem annyira ismert és a tünetei sok mindennel összetéveszthetőek. Általában a szülők már korábban érzékelik, hogy baj van, SMA-1 esetében például mikor nem fordul meg a gyerek, de neurológushoz sokszor csak akkor jutnak el, amikor az orvos által javasolt korai fejlesztés se hoz eredményt, sőt a korábban megtanult mozgások is leépülnek. Emellett sok más hasonló tüneteket okozó mozgásfejlődés lemaradással járó kórkép van, és az idő egyre megy, míg végül esetleg felmerül az SMA gyanúja. Maga a betegség egyébként könnyen diagnosztizálható egy egyszerű genetikai vizsgálattal, de az időfaktor nagyon lényeges mert egy idő után a lemaradások már nem visszafordíthatók, és a jelenlegi orvosi álláspont szerint ezeken már a kezelés sem segít. Megoldást az újszülöttkori szűrés jelenthetne, de erre egyelőre még várni kell.
„KEDVES SZÜLŐK, ZENTE TÚL FOGJA ÉLNI ÖNÖKET!”
Az SMA korábban gyógyíthatatlan betegségnek számított, az SMA-1,2-ben szenvedő gyerekek általában már a 2 éves kort sem érték meg. Ezért is számított hatalmas előrelépésnek, hogy 2017 júniusában engedélyezték az egész Európai Unióban, így Magyarországon is a nuszinerszen hatóanyagú Spinraza készítmény alkalmazását.
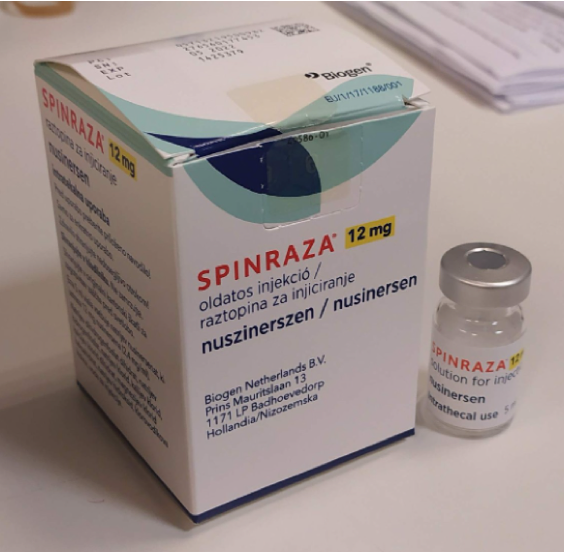
Ez a gyógyszer az SMN-2 génre hat, ennek serkentésével növeli az SMN fehérje mennyiségét a szervezetben, ezáltal megállítja a hanyatlást, és megfelelő kiegészítő terápiák mellett kismértékű fejlődést is lehetővé tesz. Zente például „a Spinraza előtt annyira leépült, hogy már csak a lábfejét tudta mozgatni.” „Az első dózis után 3 nappal pici változás már érzékelhető volt, 4 adag után tudott ülni.” A Spinraza kezelés sem olcsó, egy ampulla 22,5 millió forintba kerül, és a 4 telítő adag után 4 havonta kell egy fenntartó dózis, amit minden alkalommal egyedi elbírálás alapján finanszíroz a NEAK (Nemzeti Egészségbiztosítási Alapkezelő) a 18 év alatti gyerekeknek. Ráadásul a gyógyszert a gerincvelőbe adják, ami invazív beavatkozásként sok kockázattal jár, valamint számos mellékhatást leírtak. Ugyanakkor ez mégiscsak egyfajta megoldást jelent, Zente édesanyja így írt erről az egyik Spinraza kezelés utáni posztjában:
„Itt el is hangzott egy igazán sokkoló mondat, amelyet a hölgy hatalmas örömmel mondott ki, de bennem óriási visszhangja volt a szavainak: Kedves Szülők, Zente túl fogja élni Önöket! Szíven üt még most is. Hiszen ez lenne a természetes, a normális, de mivel hazánkban csak tavasz óta érhető el a kezelés, így még tavaly is azt kellett mondani a hozzánk hasonló családoknak, hogy nagyon sajnálják, legyenek sokat a babával, mert nem sokáig lesz már velük…”

(Fotó: Pinczés Alexandra)
A VILÁG LEGDRÁGÁBB GYÓGYSZERE
A Spinrazánál előbb leírt problémák világossá teszik, hogy miért is nehézkes a hosszútávú alkalmazás, épp ezért hatalmas áttörést jelentett 2019 áprilisában, amikor az FDA (amerikai Élelmiszer- és Gyógyszerengedélyezési Hivatal) bejelentette, hogy úttörő terápiaként engedélyezik a klinikai vizsgálatok 1. fázisában tartó Zolgensma törzskönyvezését. A Zolgensma génterápiás gyógyszer, amely egy adeno-asszociált vírus vektor segítségével juttatja be a hiányzó génszakaszt a szervezetbe, és a várakozások szerint csak egyetlenegyszer kell alkalmazni. Mindez szakmai szempontból hatalmas (bár számos vitát kiváltó) lépés, a közvéleményt mégsem ez, hanem a gyógyszer csillagászati – 700 millió forintnak megfelelő amerikai dollár – ára ragadta meg. Sokakat fel is háborított ez az összeg, azt azonban tudni kell, hogy a Zolgensma, egy telje
sen új, innovatív gyógyszerforma, amelynek nincs mihez hasonlítani az árát. A mögötte álló rengeteg kutatásnak-fejlesztés költségét pedig egy öt éves szabadalmi oltalom ideje alatt kell „behozni” a gyógyszercégnek. A gyógyszer ára a világ minden országában ugyanannyi (ez sem jellemző egyébként) és egyelőre nem néz ki úgy, hogy csökkenteni fogják. A gyógyszergyártó egyetlen „engedménye” hogy úgynevezett gyógyszerlottót indított, amelyre a beteg gyerekek szülei pályázhatnak, és meghatározott időszakonként kisorsolnak 1-1 gyereket, aki megkaphatja a gyógyszert.
Életmentő milliók egy tálcán (Fotó: Magyar Bernadett)
A gyógyszer nagyszerű újdonsága mellett, a fázis 1 utáni törzskönyvezés azt jelenti, hogy mindössze 15 gyereken vizsgálták a Zolgensma hatását az engedélyezés előtt, közülük 11 képes lett ülni, kettő fel tud állni, kettő járni tud és egy sem szorult gépi lélegeztetésre. Nem csoda hát, hogy az orvostársadalmat megosztotta a kérdés: viszonylag biztonságos, de invazív Spinraza kezelés, vagy az új noninvazív géntechnológia, ami szinte egy ismeretlenbe való ugrással ér fel.
Európában Zente előtt 2 portugál és 1 francia kisgyereket kezeltek a gyógyszerrel, a magyar kisfiú tehát a negyedik ampullát kapta meg a Bethesda Gyermekkórházban. A gyógyszer alkalmazásánál nem csak az összeg előteremtése jelentett kihívást, a kezelésre vállalkozó kórháznak rengeteg szakmai, jogi, logisztikai problémával kellett szembenéznie, amelyekből azóta tapasztalatot kovácsoltak. Kérdések azonban még mindig vannak, főleg arról, hogy mit hoz a jövő… Mindezekről a következő részben olvashattok.
Zente facebook-odala: Zente SMA 1 Tiny Hero – Stronger than SMA
További források:
https://www.zolgensma.com/
https://www.ema.europa.eu/en/documents/overview/spinraza-epar-summary-public_hu.pdf