A recent publication in Nature Genetics presents findings on the hereditary aspects of nephrosis – a disease leading to renal failure – which has substantially changed conventional understanding of the disease. The publication’s leading author, Dr. Kálmán Tory is an assistant professor at Semmelweis University’s 1st Department of Paediatrics.
 The publication is the result of the collaboration between two academic research groups at the Department of Paediatrics (an MTA-SE Paediatrics and Nephrology Research Group headed by Dr. Tivadar Tulassay and the MTA-ELTE Protein Modelling Research Group headed by Dr. András Perczel) and a laboratory in Paris (INSERM U983 laboratory).
The publication is the result of the collaboration between two academic research groups at the Department of Paediatrics (an MTA-SE Paediatrics and Nephrology Research Group headed by Dr. Tivadar Tulassay and the MTA-ELTE Protein Modelling Research Group headed by Dr. András Perczel) and a laboratory in Paris (INSERM U983 laboratory).
[pullquote]We can tell with most of the combinations whether it’s faulty or not, therefore most of the parents who has been considered endangered until now can feel relieved[/pullquote]Dr. Kálmán Tory told our website that observations made next to the sickbed provided the point of departure for the research and the results of which will be put into practice at the sickbeds as well. The paediatrician observed that if the ordinary (aka Mendelian) inheritance prevailed in a recessive nephrosis syndrome then there should be considerably more patients suffering from the disease. The efforts aiming at explaining this observation resulted in the discovery of the new hereditary model. According to their results, even if both parents’ gene copies are faulty, their child might not necessarily be sick. The child will only inherit the disease if certain defective traits are coupled with certain defective traits since it has great relevance “who is in pair with who”. In other words, it matters which maternal defects pair up with which paternal defects. “We can tell with most of the combinations whether it’s faulty or not, therefore most of the parents who has been considered endangered until now can feel relieved” – according to Dr. Kálmán Tory who added that these findings may change the genetic counselling in case of other recessive hereditary diseases as well.
Consequently this means that the authors observed a genetic polymorphism the pathogenicity of which is conditional upon the coupling mutation: certain mutations lead to disease while others coexist “peacefully”. This is similar to the classic Mendelian experiment where we would analyse a gene’s four alleles (A, B, C and D) two of which (A, B) would produce green, while the other two (C, D) would produce yellow pea pod in homozygous state. The discovery is that the A and B alleles behave differently in contrast to C and D: while A shows dominance in both cases, B shows recessive behaviour in one case. This phenomenon may change the understanding of the pathogenicity of polymorphisms since, due to the newly discovered hereditary process, other frequent polymorphisms may also be responsible for the appearance of rare monogenic diseases if they are coupled with the “right” mutation.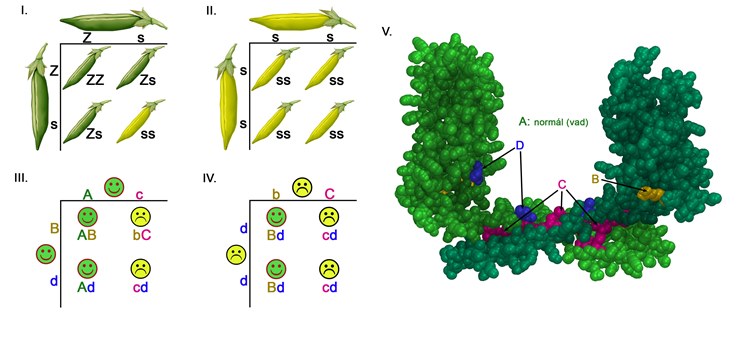
The summary of the publication, which was created and sent to us by the Hungarian authors, shows clearly that the newly discovered hereditary process and its consequences were analysed and proven on four different levels:
- On an epidemiological level, they pointed out the contradiction between the risk stemming from the classic hereditary process and the observed epidemiological fact, in other words, the divergence between the frequency of the polymorphism allele and the prevalence of the disease.
- In cooperation with the French laboratory, they managed to identify healthy family members in participating families who were thought to have the pathogenic genotype but were not sick nevertheless.
- Clinical observations were successfully proved on cell level through the analysis of the localisation of podocin protein within the cell coded by the analysed gene. The podocyte cells and the transfected podocyte culture obtained from the patients’ urine showed that in case the analysed polymorphism couples with certain mutations then the coded protein, the podocin, cannot get to its designated destination – to the cell membrane – and this leads to damaged filtration functions of the kidneys.
- Finally the employees of ELTE, Dóra K. Menyhárd, Pál Stráner and András Perczel correlated the consequence of genetic modification with the inclination of the podocin protein to form dimers on molecular and even on atomic levels. Thus, they explained the differing effects of polymorphism mutation couples through changes in protein space structure. The analysis showed that the change in the podocin protein’s ability to couple is behind the phenomenon. In case if one of the monomers is affected by the analysed polymorphism, while the other one is distorted by one of the specific coupling mutations then the dimer’s structure will become abnormal. This leads to the change of subcellular localisation!
Apart from extending the traditional hereditary (Mendelian) rules, the outstanding contribution of the publication is that it involved an unusually high variety disciplines. The formulation of the problem was honed through several stages: starting from the observations made at the sickbed, through the localisation of the subcellular protein all the way to the identification of its space structure characteristics.
The tackling of the problem is the result of the intensive cooperation of two University laboratories (MTA-SE and MTA-ELTE) as well as a Paris laboratory. Dr. Kálmán Tory, the Bolyai scholarship paediatrician was the main initiator behind the project which was launched thanks to his perseverant efforts. The cooperation between the French and the two university laboratories, which led to the identification of the gene-decoding protein after a decade and a half, demonstrates clearly the possibilities which underlie cross-border scientific efforts.
Pálma Dobozi
Translated by: Bonifac Makkai
Photo: Attila Kovács, SE
Tory K, K Menyhárd D, Woerner S, Nevo F, Gribouval O, Kerti A, Stráner P, Arrondel C, Tulassay T, Mollet G, Perczel A, Antignac C: Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome
Explanation of the diagram
Difference between the traditional and the mutation-dependent autosomical recessive hereditary processes
I-II. According to the traditional Mendelian hereditary process the crossbreeding of two dominant phenotype green pea pods will result in recessive phenotype yellow pea pods in maximum 25% of the cases.
III-IV. During mutation dependent recessive hereditary processes it is possible that two healthy parents with dominant phenotype (III.) and the two ill parents (IV.) have a 50-50% chance to have a child who will be sick as well. The reason behind this is that the B and C alleles’ dominant/recessive character is dependent upon the coupling allele. While the B variant is recessive in character in comparison to the C alleles (III. a sick child with ‘bC’ genotype), it remains dominant towards the D alleles (IV. healthy children with ‘Bd’ genotype). Similarly, the C allele which is dominant towards B allele is recessive in comparison to the wild A allele (III. healthy parent with ‘Ac’ genotype). [A and B alleles cause healthy phenotypes while C and D alleles cause ill phenotypes in homozygote form]
V. The void-fill model of the podocin protein’s dimer where the two monomers coded by a wild A allele are light and dark in colour. The B, C and D letters on the III-IV diagram mark the points where the changes of the amino acids are coded by B, C and D alleles. The B variant is pathogen with those (C) mutations that code the amino acids which play a role in the dimerization.