Epilepsy Research 150 (2019) 95-105
DOI: 10.1016/j.eplepsyres.2018.11.008
PéterHalásza, RóbertBódizsb, Przemysla PéterUjmab, DánielFabóa, AnnaSzűcsa
a National Institute of Clinical Neuroscience, Amerikai út 57. Budapest, H-1145, Hungary
b Semmelweis University, Institute of Behavioral Sciences, Nagyvárad tér 4, Budapest, H-1089, Hungary
Highlights
NREM sleep has major role in brain plasticity.
Interictal epileptic discharges (IEDs) couple with sleep slow waves and spindles.
The epileptic derailment of sleep networks leads to sleep related system epilepsies.
Most sleep-related epilepsies harm cognitive functions hindering brain plasticity.
Pharmacotherapy is armless against the cognitive loss driven by IEDs.
Abstract
The aim of this review is to summarize and discuss the strong bond between NREM sleep and epilepsy underlain by the shared link and effect on brain plasticity. Beyond the seizure occurrence rate, sleep relatedness may manifest in the enhancement of interictal epileptic discharges (spikes and pathological ripples). The number of the discharges as well as their propagation increase during NREM sleep, unmasking the epileptic network that is hidden during wakefulness. The interictal epileptic discharges associate with different sleep constituents (sleep slow waves, spindling and high frequency oscillations); known to play essential role in memory and learning.
We highlight three major groups of epilepsies, in which sleep-related plastic functions suffer an epileptic derailment. In absence epilepsy mainly involving the thalamo-cortical system, sleep spindles transform to generalized spike-wave activity. In mesio-temporal epilepsy affecting the hippocampal declarative memory system, the sharp wave ripples derail to dysfunctional epileptic oscillations (spikes and pathological ripples). Idiopathic childhood epilepsies affecting the perisylvian network may progress to catastrophic status electricus during NREM sleep. In these major epilepsies, NREM sleep has a pivotal role in the development and course of the disorder. Epilepsy is born in-, and exhibits its pathological properties during NREM sleep. Interictal discharges are important causative agents in this process.
Keywords: Brain plasticity, NREM sleep, Interictal epileptic activity, System epilepsy, Epileptic derailment, Sleep-related epilepsy
“Plasticity is defined as activity-dependent alteration in the strength of connection among neurons, a mechanism through which information is stored” (M. Steriade)
- Introduction
NREM sleep facilitates both interictal and ictal epileptic phenomena, while REM sleep suppresses them and wakefulness may allow their presence, as shown in several animal and human studies (Dinner and Lüders, 2001; Mendez and Radtke, 2001; Bazil, 2002; Foldvary-Schaefer and Grigg-Damberger, 2006). The reason why NREM sleep facilitates epilepsies, has remained unknown.
In this review, we elaborate on some basic features of the relationship between NREM sleep and epilepsy, and present the relevant research-lines for bringing a solution of this riddle closer. Contrasting the traditional seizure-oriented view, we focus on the role of interictal activity
- The plastic functions of NREM sleep
One of the outstanding achievements of recent sleep research is recognizing the essential impact of sleep slow waves both in homeostatic sleep regulation and in brain plasticity (memory and learning). According to the synaptic homeostasis hypothesis (Tononi and Cirelli, 2003, 2006), the input of the homeostatic regulation of sleep slow wave activity is the net synaptic strength deriving from the long term potentiation (LTP) processes accumulating prior to sleep (Kattler et al., 1994; Huber et al., 2006; Vyazovskiy et al., 2000). The homeostatic increase of slow waves holds also true for regional, localization-related tasks The activity of specific brain regions during wakefulness leads to increased sleep slow wave activity in that region (Kattler et al., 1994; Huber et al., 2006). As the frontal lobes are deeply involved in the regulation of behavior and cognition, regional sleep homeostasis is best shown by the abundance of sleep slow waves over the frontal lobes in adult human subjects (Horne, 1993; Cajochen et al., 1999; Finelli et al., 2001). One of the main assumptions of the synaptic homeostasis hypothesis is that central nervous system synapses are downscaled during deep NREM sleep due to the rhythmic slow oscillation predominating this phase. The decrease of slow waves across the night sleep reflects this synaptic downscaling enabling synapses to participate in the plastic process again (Tononi and Cirelli, 2006).
Due to intensive plastic changes, there is high synaptic potentiation in early childhood, resulting in an abundance of sleep slow wave activity (“deep sleep”). Contrarily there are less sleep slow waves in the elderly (“superficial sleep”) (Colrain et al., 2010), although high levels of cognition might dampen this age-related decrease of slow waves (Pótári et al., 2017). Sleep deprivation results in the well-known prefrontal-type cognitive syndrome, caused by the lack of opportunity for synaptic downscaling (Durmer and Dinges, 2005). A sleep deprivation-related prefrontal syndrome may occur in insomnia, sleep apnea (Hauri, 1997; Boufidis et al., 2004; Altena et al., 2006; Puertas et al., 2004), and in other conditions sharing the loss of NREM sleep (Simor et al., 2011).
The exclusive role of sleep slow waves in plastic changes through the downscaling process has recently been challenged (Frank, 2012; Grosmark et al., 2012). The original synaptic homeostasis hypothesis of Tononi and Cirelli has pointed out that the exponential decline of sleep slow wave activity parallels the refreshment (downscaling) of synapses potentiated by the cognitive activity performed in pre-sleep wakefulness. They claimed that this downscaling is a global function of slow wave sleep. However, research on memory consolidation (see in chapter 5.2) has shown that synaptic potentiation (upscaling) occurs within the hippocampo-frontal brain circuitry during slow wave sleep (memory consolidation), too. Therefore NREM sleep plays an important double role participating both in the down- and up-scaling of synapses (Niethard et al., 2017)
In addition to slow waves, also sleep spindles participate in the sleep-related plastic process. Sleep spindles are tightly coupled with synaptic potentiation (Ulrich, 2016). They coalesce with other NREM sleep oscillations. They are enveloped by the up-states of the slow (<1 Hz) oscillations and group with ripples (Clemens et al., 2011). The coupling and fine-tuning of these three oscillations (slow waves, spindles and ripples render the cortex receptive for plastic changes (Born et al., 2006, Diekelmann and Born, 2010; Mölle and Born, 2011; Mölle et al., 2011).
The increase of spindle density and the length of N2 sleep highlight the role of sleep spindling in memory formation after visuospatial or verbal learning (Staresina et al., 2015; Fogel and Smith, 2006; Siapas and Wilson, 1998). Gais et al. (2002) have shown that after intensive declarative memory tasks, frontal spindling density increased during the first 90 min of sleep. Schabus et al. (2004) comparing those subjects who increased their spindle activity after learning with those who have not, found that the increase of spindle activity correlated with post-sleep performance. Several studies confirm the role of spindling in the protection of engrams in the memory consolidation process (Diekelmann and Born, 2010, 2010; Payne and Kensinger, 2011; Lewis and Durant, 2011) as well as their involvement in the integration of information with the stored memories (Tamminen et al., 2010).
NREM physiology links with the burst-firing working mode of the thalamo-cortical system, liberated by the inhibition of the arousal system while falling asleep. The appearance of spindling and slow waves as characteristic by-products of the cortical pyramidal cells, thalamic relay neurons and the reticular nucleus of the thalamus (NRT) reflects this liberation (Steriade, 2003a) (Fig. 1).
Fig. 1.
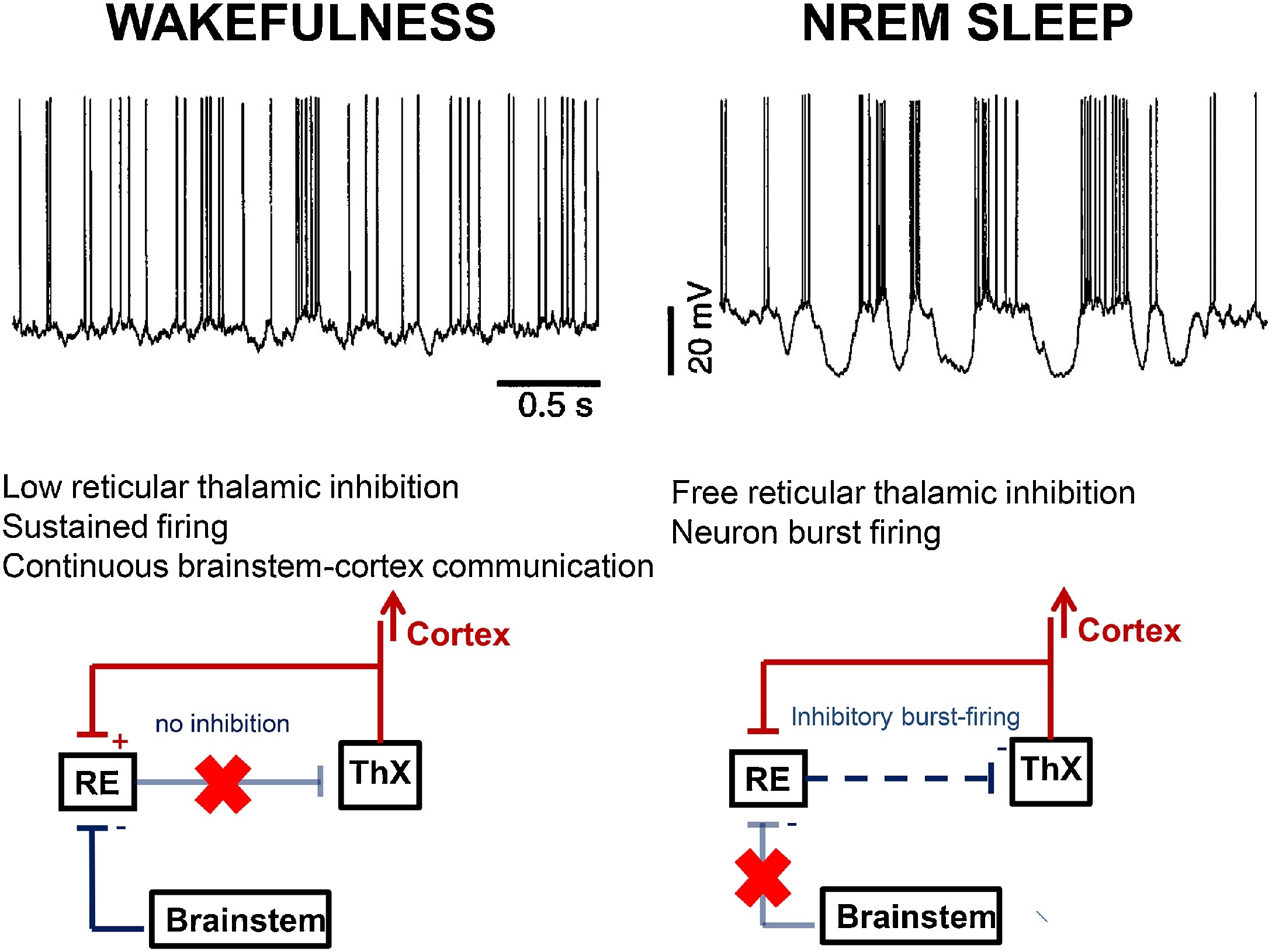
Differences in the thalamo-cortical circuitry in wakefulness and NREM sleep. During wakefulness (left) brainstem cholinergic innervation through the thalamic relay cells (ThX) keeps connection between the environment and the cortex, indicated by the continuous tonic activity (top row left) in the cortical cells (excitation red, inhibition blue). The brainstem cholinergic innervation inhibits the thalamic reticular nuclei (RE) (pale blue line with red X), therefore the reticulo-thalamic inhibition is inactive. During NREM sleep (right) the cholinergic inhibition of the reticular nuclei does not work (pale blue line with red X). Therefore the reticular nuclei provide inhibitory impulses to the thalamic relay cells (blue dashed arrow). Thus the thalamo-cortical impulse flow is interrupted and a burst-firing mode (top row right) dominates; producing sleep spindles and slow waves.
During wakefulness, the thalamo-cortical system works as a relay center, conveying external input towards the cortex. This is executed by the so-called “tonic activity” of the network reflected by a desynchronized (low amplitude fast) EEG pattern. Upon sleep-onset, the thalamo-cortical system changes to an excitatory-inhibitory cycle, referred to as burst-firing working mode. In this mode, the NRT periodically inhibits the firing of the thalamic relay nuclei. This is as reflected by cortical spindles or delta waves, depending on the relay cells’ level of membrane polarization (Steriade, 2003b). The NRT produces spindling even after isolation from rest of the thalamus (Steriade et al., 1985).
The sleep-related enhancement of declarative memory consolidation does not exclusively rely on the enhanced plasticity provided by sleep spindling and slow waves in the thalamo-cortical system. The hippocampo-neocortical dialogue and the sharp-wave ripples (SPW-Rs) are concomitant patterns, primarily involved in the strengthening of memory traces and their transfer from the hippocampus to neocortical networks. The active systems consolidation theory (Rasch and Born, 2013) states that memory traces, initially stored in hippocampal circuits, are transferred to neocortical networks in NREM sleep via the interplay of slow waves and SPW-Rs both prevailing during these states.
SPW-R is a robust, far reaching population episode, the most synchronized event of the mammalian brain (Buzsáki, 2015), elevating the excitability of the hippocampus and connected structures (Buzsáki, 1986; Chrobak and Buzsáki, 1998; Csicsvári et al., 1999). It is detected in isolated hippocampal slides as well. The ripple is a high frequency (200 Hz in animals, 150 Hz in humans) event of the CA1 pyramidal layer of the hippocampus, evoked by the sharp wave originating in CA3 (Buzsáki, 2015). It is a fast oscillatory response to the sharp wave, remaining local between the CA3 pyramidal cells and the peri-somatic inhibitory neurons (Schlingloff, et al., 2014; Gulyás and Freund, 2015). SPW-R can elicit LTP; which in turn changes the synaptic weights between the hippocampal neurons. The duration of ripples in rat is 30–150 ms and the amplitude does not exceed 2.5 mV; the spike amplitude is higher, the duration is shorter and the coupled ripples are more synchronous in humans. A substantial amount of the hippocampal neurons (50–150,000 neurons) participate in it.
The elimination of SPW-Rs obstructs memory consolidation (Girardeau et al., 2009; Roux et al., 2017). Several studies have shown that the cellular spike sequences in slow wave sleep are identical with the sequence observed during SPW-Rs just faster and compressed (Foster and Wilson, 2006; Ji and Wilson, 2007; Nádasdy, 2000). This mechanism has not been demonstrated in humans, but the increase of SPW-Rs after learning supports the existence of a similar effect (O’Neil et al., 2000; Cheng and Frank, 2008).
- The epileptic derailment of NREM sleep plastic functions
Taxonomical approach has shown epilepsy to be a heterogeneous disorder with different lesional and genetic etiologies, widely different symptoms and various semiology. Despite this diversity, there is a growing belief in a shared pathomechanism. This belief is supported by the recognition of high-frequency epileptic oscillations (HFO) which have recently become a candidate for a universal marker of epileptogenicity (Zijlmans et al., 2012; Frauscher, et al., 2017).
Epilepsy is a chronically increased excitability state of certain brain structures (essentially in the archi-, and neocortex) resulting in a proneness to generate epileptic seizures. Buzsáki (Buzsáki, 1986) remembered the work of Goddard and Douglas, 1975 first supposing that the plastic processes of memory engram-formation and epileptogenesis are similar. The repetitive stimulation of a neuron may generate physiological LTP in a synaptically connected second neuron, resulting in engram-formation. Similarly, in epileptic kindling, the enduring stimulation of epilepsy-prone brain structures leads to after-discharges and results in spontaneous seizures later. If epilepsy starts in early life at the time of intensive processes, the epileptic over excitation may change the functions of certain brain systems, based on the Hebbian „firing together, wiring together” principle and take over their regulation. Beenhakker and Huguenard (2009) have called this pathological learning process, leading to the development of epilepsy, the „hijacking” of certain brain systems. However, catchy, this metaphor rests on the incorrect idea that epilepsy is a foreign outside force raping the brain. The assumption that epilepsy would be a derailment of inherent plasticity, seems more plausible. Viewing epilepsy as a derailment of normal plastic functions would explain its high prevalence and make it the by-product of the ability to learn.
The cognitive harm of abundant IEDs even without any seizures in EES/LKS has been evidenced (Tassinari et al., 2009). The large scale cognitive deficits recognized increasingly in idiopathic focal childhood epilepsies (Baglietto et al., 2001; Neuman et al., 2016; Wickens et al., 2017) support the cognitive harm of IEDs, too the link of hippocampal spiking with a special cognitive deficit restricted to memory impairment has been increasingly accepted as well (see in Chapter 5.2.1).
Several studies have shown that NREM sleep slow oscillations and spindling fuel memory consolidation and synaptic homeostasis. What is the evidence for sleep oscillations and epileptic discharges relying on similar mechanisms?
During NREM sleep the amount, quality, extension and bilateral synchrony of IEDs vary along a continuum in sleep related epilepsies. The repetition rate of the discharges as well as their propagation network increase during NREM sleep, while their basic localization and morphology remains preserved. Sleep apparently unmasks the existing epileptic networks. We propose to change our view from assuming that epilepsies or at least those with a strong relation with NREM sleep are “activated” during NREM sleep, to one recognizing that epilepsies are born and work during sleep. Diagnostic sleeps studies do not “activate”, rather uncover epilepsy revealing every night’s NREM sleep-related epileptic events. Sleep deprivation increasing homeostatic sleep pressure makes an exception: the elevated homeostatic pressure due to sleep deprivation increases sleep pressure making an additional effect (“activation”) on top of normal night sleep on IEDs; resulting in additional augmentation of epileptic activity.
- IEDs couple with sleep EEG oscillations serving plastic functions
The distribution of IEDs is not homogenous in NREM sleep. Based on the studies of the Parma sleep research school, spikes are under the same homeostatic control as sleep slow waves. They occur frequently in the first sleep cycles and on the descending slopes within cycles (Terzano et al., 2005). In the majority of epilepsies, there is a close link with the CAP A1 phases (Parrino et al., 2000). Rolandic epilepsy (RE), Panayiotopoulos syndrome (PS); their malignant transformations – electrical status epilepticus in sleep (ESES) and Landau-Kleffner syndrome (LKS) – make exceptions, as their IEDs associate with sleep spindles (Fig. 2) (Nobili et al., 1999). This coupling of IEDs with spindles is typical in a broad class of epilepsies referred to as “perisylvian network epilepsies” (Halász et al., 2005). This interesting dichotomy – coupling with either sleep slow waves or spindles – has remained un-interpreted.
Fig. 2.
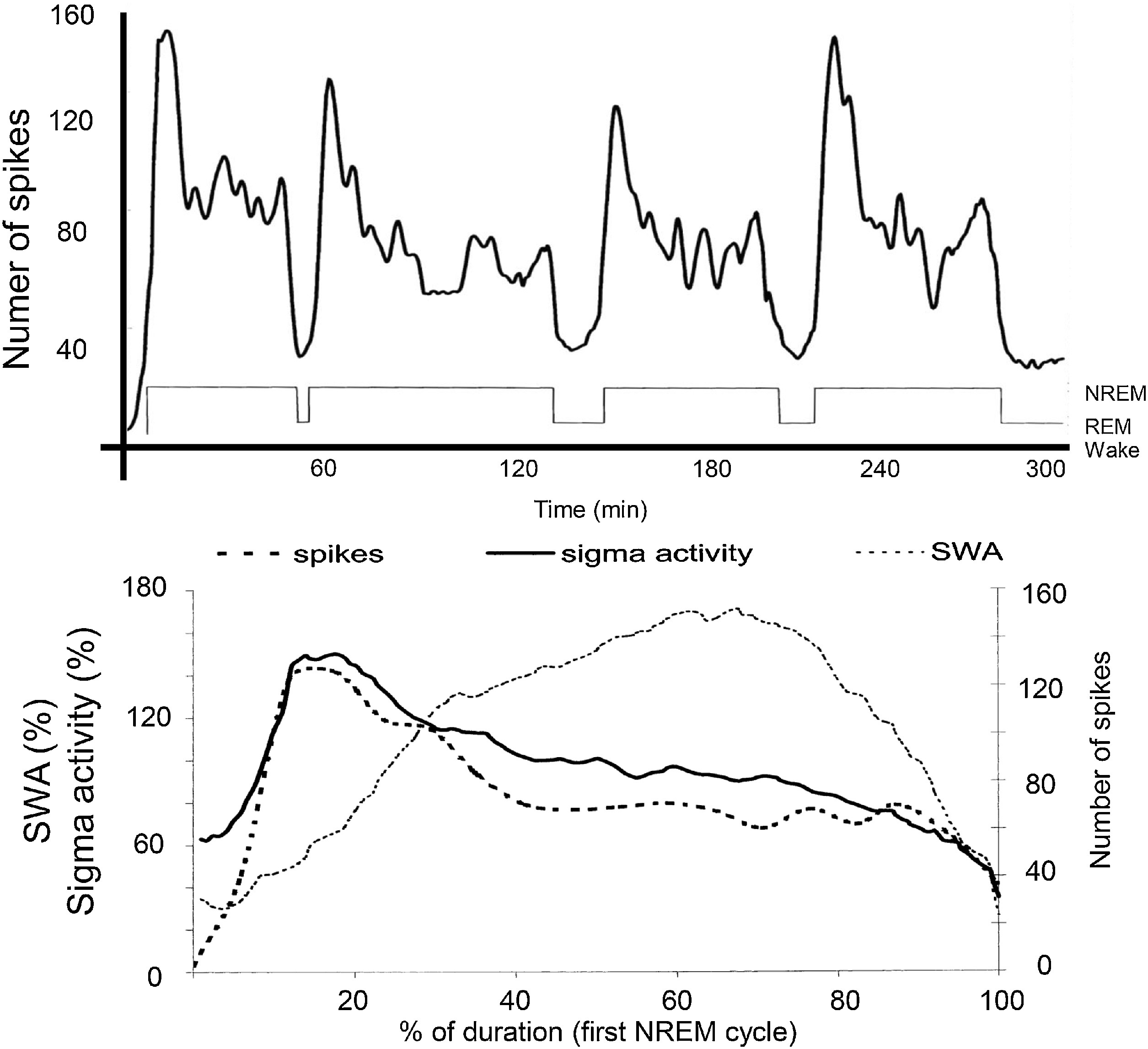
The relationship between sleep oscillations and the occurrence rate of spikes in the spectrum of perisylvian epileptic network disorders. Top: Distribution of spikes across the sleep cycle. Bottom: Distribution of spikes (dashed line; the EEG sigma band (SFA = 12–16 Hz) – continuous line- and slow wave activity (SWA = 0.5–4.0 Hz) – dotted line – across the first NREM cycle. It is clearly seen that spikes go parallel with sigma frequencies (sleep spindles), and not with SWA (After Nobili et al 2011).
Invasive presurgical evaluations in epilepsy provided more insight into the relationship between IEDs and sleep oscillations. Such links suggest a deep, much more than incidental, link of NREM sleep elements with epileptic discharges.
Frauscher et al. (2015b) have shown that IEDs and HFOs preferentially occur during high amplitude slow waves, especially during the transition from the ‘up’ to the ‘down’ states of the slow waves (unlike physiological oscillations preferring the “up” states). Our group (Ujma et al., 2014) has published congruent results showing that cortical spikes, captured by subdural electrodes in epilepsy patients undergoing presurgical evaluation preferentially occur during CAP A1 phases, suggesting the role of slow waves anchoring spikes. Additionally Ujma et al. (2017) found that the IEDs occurred during the initial down states of slow waves, and more frequently during larger slow waves. Because several studies have shown correlations between CAP A1 events and cognitive performance, the link between IEDs and sleep plastic functions through CAP A1 events can be congruently assumed.
In sum, there is a consistent participation of sleep spindles, slow waves and high frequency oscillations in sleep plastic processes. The same NREM constituents seem to link with IEDs as well, indirectly indicating that IEDs may affect sleep plastic functions.
Another aspect may further alight the importance of the link between sleep oscillations involved in plastic changes and IEDs. It is the old unsolved riddle of sleep deprivation activating epileptic manifestations. Because sleep slow wave economy is under homeostatic regulation, sleep deprivation increases the sleep pressure expressed in slow wave augmentation. The explored strong link between CAP A1 slow waves and IEDs may explain the well-known activating effect of sleep deprivation on epileptic manifestations.
- The epileptic transformation of certain physiological brain systems and the role of NREM sleep
Epileptic transformations in sleep-related epilepsies occur in at least three major physiological brain networks: the cortico-thalamic system; the hippocampo-frontal memory system and the perisylvian cognitive network.
5.1. The thalamo-cortical system and absence epilepsy
The thalamus (Williams, 1965) and the cortico-thalamic system (Jasper and Droogleever-Fortuyn, 1946) have long been related to the pathomechanism of absence epilepsy and its EEG substrate the bilateral synchronous spike-wave (SW) pattern.
Absence epilepsy is conventionally considered a product of the waking state. However, long-term video-EEG observations (Stevens et al., 1971) showed that spike-wave paroxysms prevail during transitory drops of vigilance in waking (Niedermeyer, 1972; Passouant et al., 1974; Horita et al., 1991; Gloor et al., 1973), while they are absent during full vigilance. Spontaneous SW paroxysms are inhibited by sudden increases of vigilance (Li et al., 1952; Rajna and Lona, 1989), arousal (e.g. being called by name) (Halász, 1982) and experimental stimulations of the reticular arousal system. Also REM sleep inhibits absences, and although interictal spike-wave fragments are present or even enhanced in deeper NREM sleep, the absence-like (longer) ictal SW paroxysms do not show up in deeper NREM sleep. Therefore, it is clear that absences are sleep related events promoted by transitional states between waking and falling asleep, as well as by superficial NREM sleep.
Pierre Gloor (Gloor, 1979) from the Montreal Neurological Institute was the pioneer of a systematic approach on the pathomechanism of absences with 3 Hz bilateral SW paroxysms. He has drawn attention to the role of the corticothalamic system (which he called „cortico-reticular”), and proposed that SWs (the EEG substrates of absences) emerge from the same circuit that normally produces sleep spindles by the “burst-firing” working mode during NREM sleep. SWs are the transformed epileptic variants of sleep spindles (Gloor, 1968). This idea has fertilized further on-going research (Kostopoulos, 2000, 2001).
Beenhakker and Huguenard (2009) dedicated their interesting paper to mechanisms by which epilepsy „hijacks” (co-opts, or uses as templates) certain sleep related circuits This theory has highlighted the relationship between sleep spindles and bilateral synchronous SWs, underlain by the epileptically hijacked “burst-firing” mode of the thalamo-cortical system in absence epilepsy (Huguenard and Prince, 1994, Mc Cormick, 2007). One of the prevailing views is that GABAA receptor blockade may promote this switch from spindling to spike-waves (Bal et al., 1995) by eliminating the normal intrareticular inhibition. Normal intrareticular GABAA receptor activity provides mutual inhibition between reticular neurons, regulating the level of the inhibition exerted on thalamic relay cells. If the output inhibition from the reticular cells is high (the intrareticular inhibition is low), the reticular to thalamic relay cell inhibition is effective, leading to an SW type hyper-synchronization (Huguenard and Prince, 1994, Sanchez-Vives et al., 1995). The pharmacologic blockade of GABAA receptors in ferret geniculate slices resulted in the transformation of spindles to paroxysmal activity: both the thalamo-cortical and the peri-geniculate neurons increased the intensity of their burst discharges and became phase-locked to a 2–4 Hz rhythm. Therefore, the shift from normal to paroxysmal activity might result from the mutual disinhibition of the peri-geniculate neurons, leading to an increase in their discharge rate and the augmentation in the postsynaptic activation of GABA B receptors in the thalamo-cortical neurons.
Huguenard and McCormick (2007) emphasized that “connections between the cortex and thalamus reinforce the thalamic oscillatory activity into larger thalamo-cortical network. The degree of synchrony within the thalamic network seems to be crucial in determining whether normal (spindle) or pathological (spike-wave) oscillation occur”. They recognized that the NRT is the key structure in the regulation of the complex excitability patterns of the thalamo-cortical circuit.
In the first years of the third millennium Steriade and co-workers performed a series of studies showing that both spindles and 10 Hz cortical or thalamic repetitive stimulations modelling spindles during slow wave sleep led to a self-sustained augmenting response with progressively increasing depolarization areas and unit firing. The elicited response from the thalamus influenced the coupled cortical area inducing activity similar to the evoked recruiting patterns, revealing strong thalamo-cortical synchrony in the induced plastic changes. During their experiments with augmenting responses, they registered a “paroxysmal type” of the self-sustained response in the cortical leads independently of the thalamic activity, even during thalamic hyperpolarization due to the ongoing reticular inhibitory impulses (Steriade and Timofejev, 2003). These experiments evidence that the physiological thalamo-cortical working mode may turn into paroxysmal epileptiform activity.
Steriade in his epoch-marking book summarized the contemporary results about the thalamo-cortical system in sleep and epilepsy – specifically, absence epilepsy (Steriade and Timofejev, 2003). He has shown in chronically implanted, freely moving, naturally awake and sleeping cats, as well as by ablation experiments that in absence-like seizures SW discharges „originate in the neocortex and are disseminated through mono-, oligo- and multisynaptic intracortical circuits before they spread to the thalamus and exhibit generalized features”. They stressed the propagation dynamics of the seizures from local to widespread confirming early observations on humans suggesting a focal onset (Niedermeyer et al., 1969; Niedermeyer, 1972; Bancaud et al., 1974). This was a complete break with the old „centrencephalic” theory of Jasper and Penfield supposing a midline deep (thalamic) source of the bilateral synchronous SW pattern (Penfield et al., 1954; Jasper, 1977).
An important line of studies on the „cortical drivers” of the SW pattern evolved in the first decade of the 20th century. Meeren et al. (2002, 2005) from the Nijmegen laboratory of experimental neurosciences, have described cortical seizure spots in rat genetic absence model. These seizure spots have quickly entrained the frontal cortex and the thalamic cortico-reticular structures into a bilateral synchronous oscillation in the form of SWs. Several interesting molecular features of the driver zones (Manning et al., 2004; Klein et al., 2004, Polak et al., 2007), as well as the successful therapeutic approach have been described later (Blumenfeld et al., 2008). In a review paper, Avoli (2012) summarized the brief history of the oscillatory roles attributed to the thalamus and cortex in absence seizures.
These animal and human studies provide strong evidence for the view that the underlying mechanisms of absence epilepsy rests on an epileptic derailment of the thalamo-cortical plasticity during the physiological shift from the waking state working mode to the burst-firing mode of NREM sleep.
5.2. The epileptic transformation of the hippocampo-frontal declarative memory network in mesio-temporal epilepsy (MTLE)
MTLE, the most prevalent adult epileptic disorder interweaves the hippocampo-neocortical system deeply involved in memory consolidation during quiet wakefulness and NREM sleep.
SPW-Rs are essential participants of the hippocampal memory function (Buzsáki, 1989). This intensive synchronous excitatory event is very near to the level of epileptic excitation, giving a hint, why the hippocampus is the most epilepsy-prone structure of the brain (Buzsáki, 2015). SPW-R is a normal counterpart to the epileptic spike-pathological ripple complex (Fig. 3). The latter differs from SPW-Rs only by its shorter duration and higher voltage (Fig. 4). Shatskikh et al (2006), stimulating the ventral commissure, could elicit large population spikes in the CA1 region of the hippocampal pyramidal neurons, and analogue spikes were detected along with the spontaneous ones. The stimulated animals suffered serious memory loss. Clemens, et al. (2011) and Kleen, et al. (2013) have evidenced the link between memory disturbances and hippocampal sleep spiking in human MTLE patients. Frauscher et al. (2015a) recording hippocampal spindles reported on negative correlation between sleep spindles and interictal spike counts. They assumed that hippocampal spiking lowered the rate of hippocampal spindles, indicating the transformation of physiological spindles to interictal spikes. This would be similar to the transformation proposed by Gloor (1968) from spindles to SWs in genetic absence epilepsy. Recently, Gelinas et al. (2016) using a rat kindling model, provided elegant experimental evidence for SPW-Rs’ conversion to spikes, and ripples’ interference with the temporo-frontal memory consolidation.
Fig. 3.
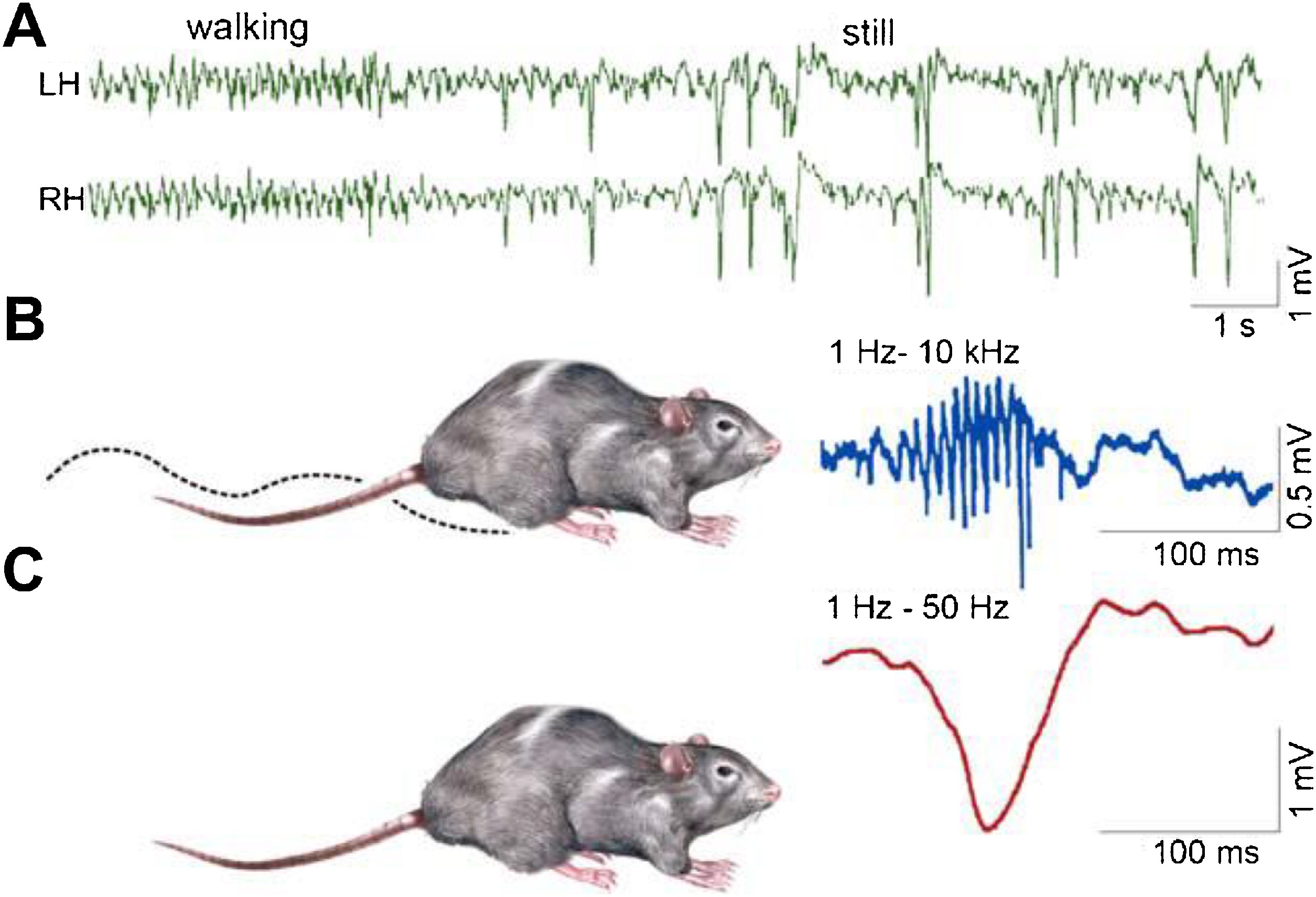
Schematic picture of sharp-waves and ripples in the rat hippocampus during the transition period from waking state to sleep. Top: EEG during active wakefulness (locomotion and orientation)- theta activity (left), while during quiet wakefulness sharp-wave-ripple complexes appear (right). Bottom: the two states of the animal (A: activity, B: quiet wakefulness. C: Enlarged picture of the complex: ripple (top) sharp wave (bottom.) (After Buzsáki, 2015).
Fig. 4.
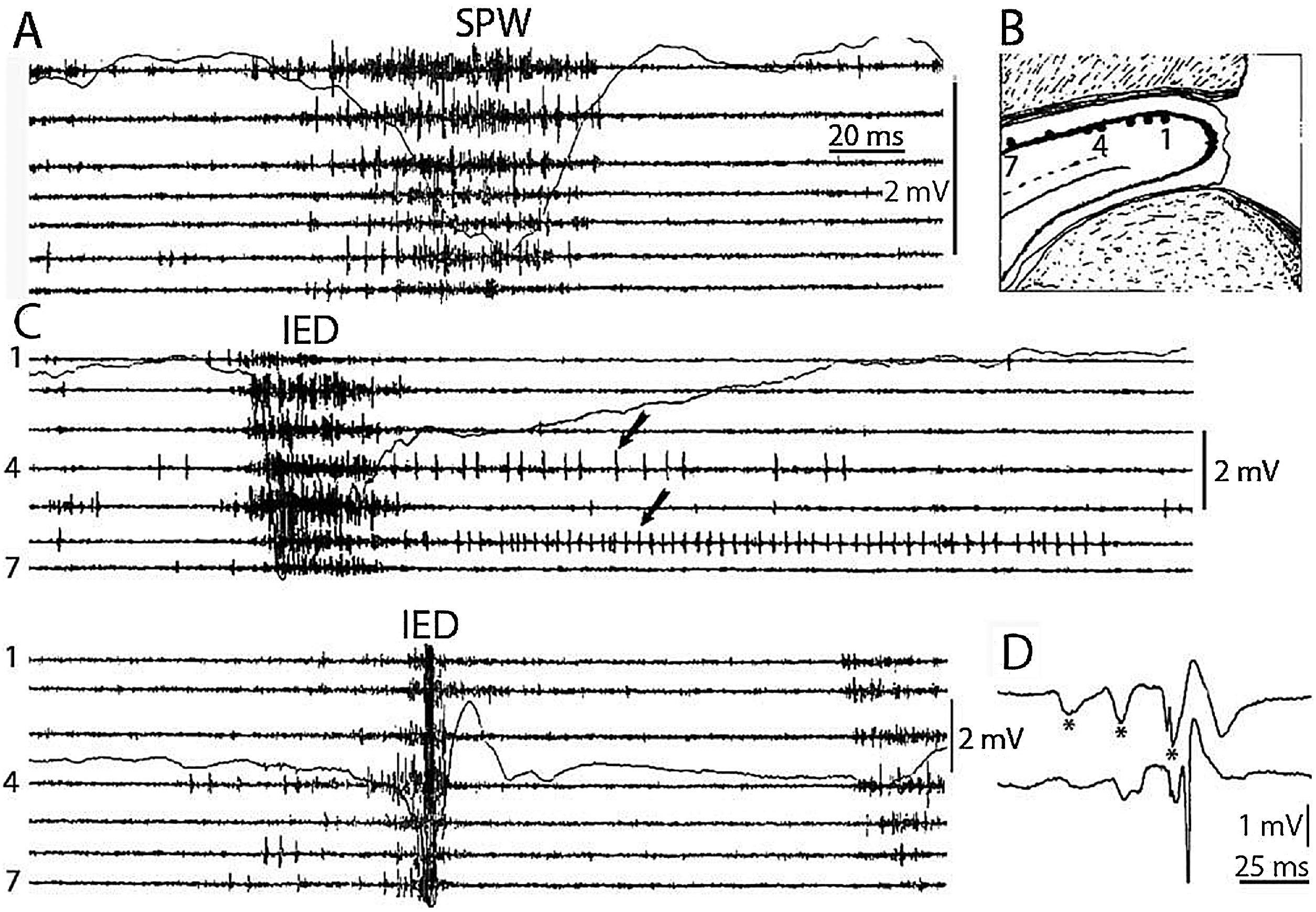
Comparison of sharp waves ripples (SPW) in the intact rat hippocampus (A) and interictal epileptic discharges (IEDs) after disconnection from its subcortical connections (C), by fimbria-fornix lesion (B). Channels represent different locations along the axis of the hippocampus (1–7) Arrows show prolonged post-IED activity in two putative interneurons. IEDs were the products of a reverberation in the enthorhinal cortex- hippocampus loop evoked by enthorhinal induced responses (asterisks) in insert (D) bottom trace represent the CA1 pyramidal layer. Reverberation was terminated by the appearance of a large population spike. See tighter synchrony of population bursts and larger amplitude of the field responses during IEDs. (After Buzsáki, 2015).
These results suggest that interinterictal spiking might contribute to the memory loss of MTLE patients by at least two mechanisms:
1 Daytime spikes behave as senseless engrams: they annex synaptic capacity; limiting the elaboration of normal engrams (this is hypothetical at present).
2 Sleep-related epileptic discharges are the dysfunctional counterparts of SPW-Rs, unable to process normal engrams (Gelinas, et al., 2016, Boly et al., 2017).
These data suggest that memory disturbances in MTLE are due not only to the static hippocampal sclerosis factor, but also to the transformation of normal hippocampal electrophysiological activity into pathological (epileptic) analogues.
5.2.1. How do clinical data reflect the impact of the hippocampal memory system’s transformation on the memory impairment in MTLE?
Since the elaboration and follow up studies of the famous H.M. case after his bilateral hippocampectomy (Milner, 2005), the essential role of the paired hippocampi in human declarative memory has become obvious. Due to the worldwide spread of the pre- and post-surgical neuropsychological testing of TLE patients as well as to the dramatic progress of neuroimaging, the understanding of the memory process has slowly grown, however, it is far from complete. The main factors limiting our knowledge are the small availability of neuropsychology capacity for non-surgical patients and the lack of long-term follow-up studies.
In MTLE, several ictal semiology-features are related to memory disturbances (e.g. déjà-vu and jamais vu feelings, pure amnestic fits etc.) and one may wonder if acute transient memory losses, rather than disturbances of consciousness, underlie the patients’ loss of contact in their partial seizures. Revealing hippocampal sclerosis as the cause of side-specific memory disturbance, we had hoped to identify a solid basis for the memory deficits of MTLE patients. However, the gradual transformation of SPW-Rs to IEDs obstructing memory formation and/or consolidation, makes an additional cause of memory disturbances (Fig. 5). Interictal spikes were shown to disrupt cognitive and memory processes independently from the seizure-onset zones (Dinkelacker et al., 2016).
Fig. 5.
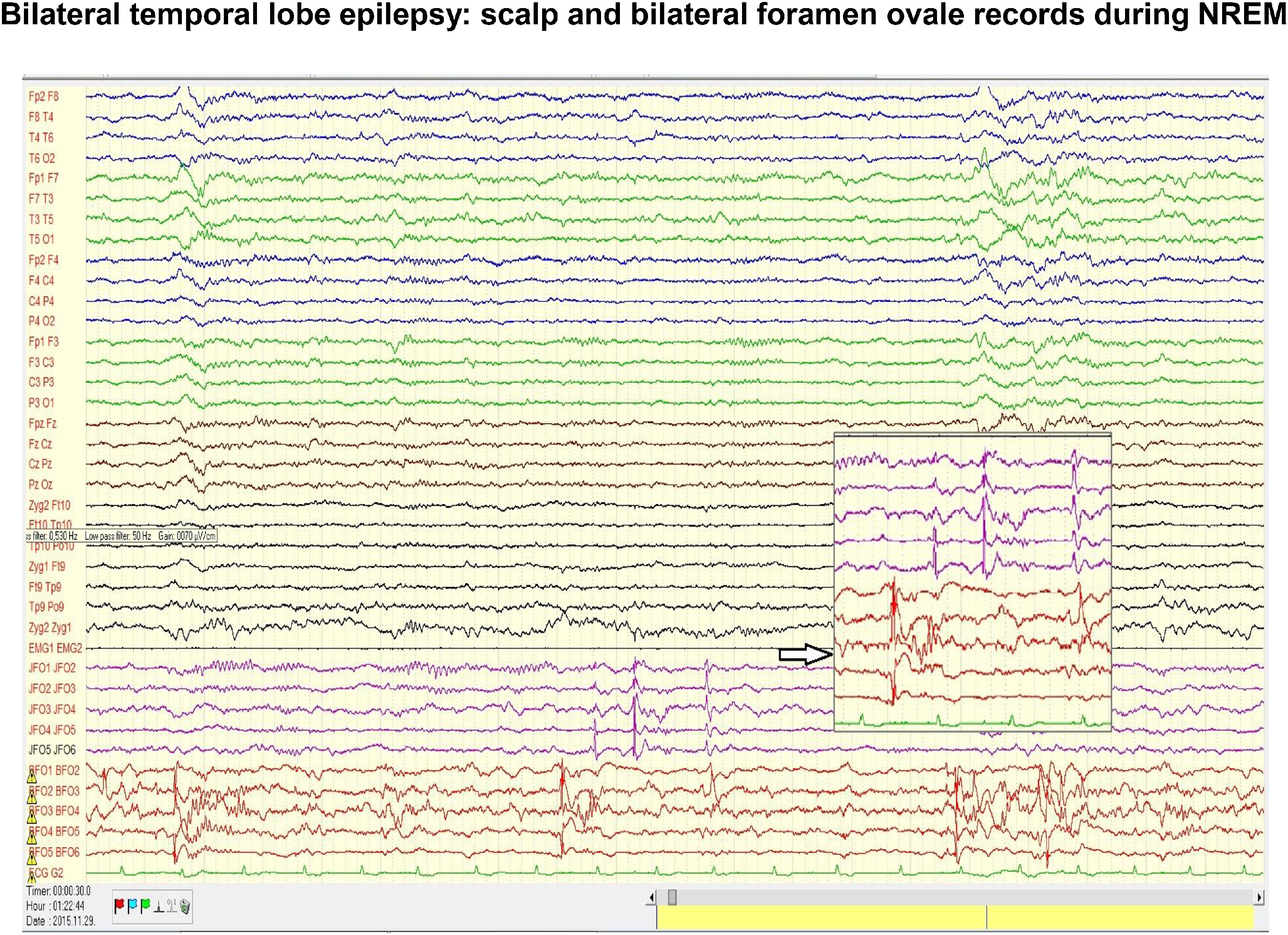
Scalp and bilateral foramen ovale (FO) records in a patient with temporal epilepsy. Note the intensive interictal spiking independently in the two foramen ovale records. In the scalp electrodes there is no trace of the flood of spikes seen in FO electrodes. The coupling of spiking with the slow groups (CAP A1 phase) registered on the scalp during NREM sleep, could be seen twice from the three clusters of spikes. Insert indicated by empty arrow enlarges the independency of the left and right FO electrodes.
5.3. The perisylvian cognitive network
The perisylvian cognitive network is a human neo-formation. It is the functional system of speech, reading/writing and working memory, closely interconnected with the thalamus. Due to its delicate functions serving human communication, it is highly vulnerable to developmental errors.
5.3.1. Idiopathic age dependent childhood epilepsies
Idiopathic age dependent childhood epilepsies represented by Rolandic epilepsy (RE) and Panayiotopoulos syndrome (PS) are classic developmental abnormalities, associated with mild cognitive dysfunctions (Wickens et al., 2017). Several researchers (Doose and Baier, 1989; Koutroumanidis and Panayiotopoulos, 1993, Panaiyotopoulos 2002) considered them as conditions of transient developmental delay with increased excitability. Interestingly, IEDs (the most prominent type being centro-temporal spikes – CTSs) also in these conditions show a high degree activation during NREM sleep.
These epilepsies are often believed “benign” with no cognitive consequences. However, in the last ten years several publications have revealed a wide range of associated cognitive and behavioral disturbances (Van Bogaert et al., 2012). A systematic review and meta-analysis of 42 case-control samples comprising 1237 still untreated BCTE children and 1137 healthy controls has shown significantly lower scores in eight cognitive domains. The largest deficit phenotypes affected long-term storage and retrieval (Wickens et al., 2017). Other studies found cognitive loss in language and academic performance (Neumann et al., 2016). Taking into consideration the level of spiking in NREM sleep, the neuropsychological loss seemed to be associated to the presence of bilateral discharges (Chan et al., 2017). At the same time, neuroimaging revealed thinner cortex in RE patients compared to controls in the frontal, temporal and occipital regions (Vannest et al., 2015), supporting the notion of a neurodevelopmental etiology.
The revealed cognitive dysfunctions fill the gap between the seemingly benign regional age dependent childhood hyperexcitability syndromes (RE and PS) and their atypical variants progressing to ESES and LKS, supporting the spectrum concept. A recent conference on idiopathic focal epilepsies: the “The lost tribe- state of the art” (Pal et al., 2016) supports this approach in a multilateral way.
5.3.2. The transition to ESES/LKS
Since the seventies, several age dependent childhood epilepsy patients with a malignant transformation have emerged in the literature: they developed severe speech-loss or suffered from global cognitive decline (Patry et al., 1971; Tassinari et al., 2000). Their IEDs flood over the cortical convexities during all NREM stages constituting electrical status epilepticus. This nearly continuous widespread bilateral spiking during slow wave sleep may last for months or years without clinical seizures. Evidence has accumulated to support a causative link between electrical status epilepticus and the severe cognitive deficit, now considered unequivocal. Tassinari et al., (1977) called it an “encephalopathy related to electric status epilepticus” (1977), now it is called electrical status epilepticus in sleep (ESES). Its treatment has remained unresolved; the usual antiepileptic drugs are ineffective.
It has been recognized that a deviant RE course may underlie ESES (Dalla Bernardina et al., 1978, 1991, Saltik et al., 2005). In the meantime, acquired epileptic aphasia, a variant of ESES underlain by abundant regional epileptiform discharges during NREM sleep, has also been described and named Landau-Kleffner syndrome (LKS), honoring its discoverers. The affected children lose speech-perception after normal speech had developed previously. Congruently, the spikes focalize in the posterior temporal region of the dominant hemisphere (Kellermann, 1978), despite the lack of an underlying lesion.
The epileptic transformation of the perisylvian communication network differs from the previously described ones. Here we cannot point out a physiological sleep oscillation transforming to an epileptic variant. These conditions are featured by CTS, not representing a pathological transformation of any sleep oscillation, it is rather a marker of cognitive impairment specific for the perisylvian region. CTS is poorly understood at the border of non-epilepsy (shown by the absence of ripples and its presence in non-epileptic patients e.g. in autism and ADHD as well) and epilepsy (associating to ripples in RE and PS) (Rossi et al., 1995; Nass and Devinsky, 1999; Silvestri et al., 2007. Danhofer et al., 2018). The infantile version of CTS behaves as an augmented evoked potential, responding to acoustic and tactile stimuli (de Marco and Tassinari, 1981; Fonesca and Tedrus, 1994). One might consider it as the EEG manifestation of delayed local cortical development and increased local excitability (Doose and Baier, 1989, Koutromanidis and Panayiotopoulos, 1993, 2002) with a potential to regress or progress to epilepsy and – sometimes – to malignant forms.
We published a conception called „The perisylvian epileptic network” on the spectrum of benign focal childhood epilepsies transforming to malignant encephalopathies as LKS or ESES (Halász et al., 2005). In line with us Di Negri (1997) has issued a similar unifying concept and Fejerman (2009) has analyzed the features of the malignant disease-process, which he named “atypical Rolandic epilepsy”.
Recent genetic research on idiopathic focal childhood epilepsies and the spectrum involving ESES and LKS supports the roles of the SRPX2 and ELP4 genes in the inheritance of epileptic sleep activation with impact on cell motility, migration and adhesion (Rudolf et al., 2009). Lemke, et al. (2013) showed changes in the gene encoding the NMDA receptor NR2A subunit as a major genetic risk factor for the whole spectrum. They have found new heterozygous mutations in GRIN2A in 27 of 359 affected individuals from two independent cohorts with increasing frequency toward the ESES end of the spectrum. In the RE-ESES spectrum (including LKS) (Lesca et al., 2012) have found increased copy number variations in the genomic architecture of several genes (encoding cell adhesion proteins) involved also in autism spectrum disorders.
Endophenotypes are shared genetic modules of phenotypically different complex disorders (e.g. schizophrenia, autism, ADHD and certain epilepsies). CTS is an endophenotype shared by RE, autism spectrum disorders and ADHD, expressing focal hyperexcitability and local/regional cognitive deficits (Fig. 6). Similarly, the critical increase in sleep activation of CTS-like IEDs might be an endophenotypic feature for the whole spectrum.
Fig. 6.
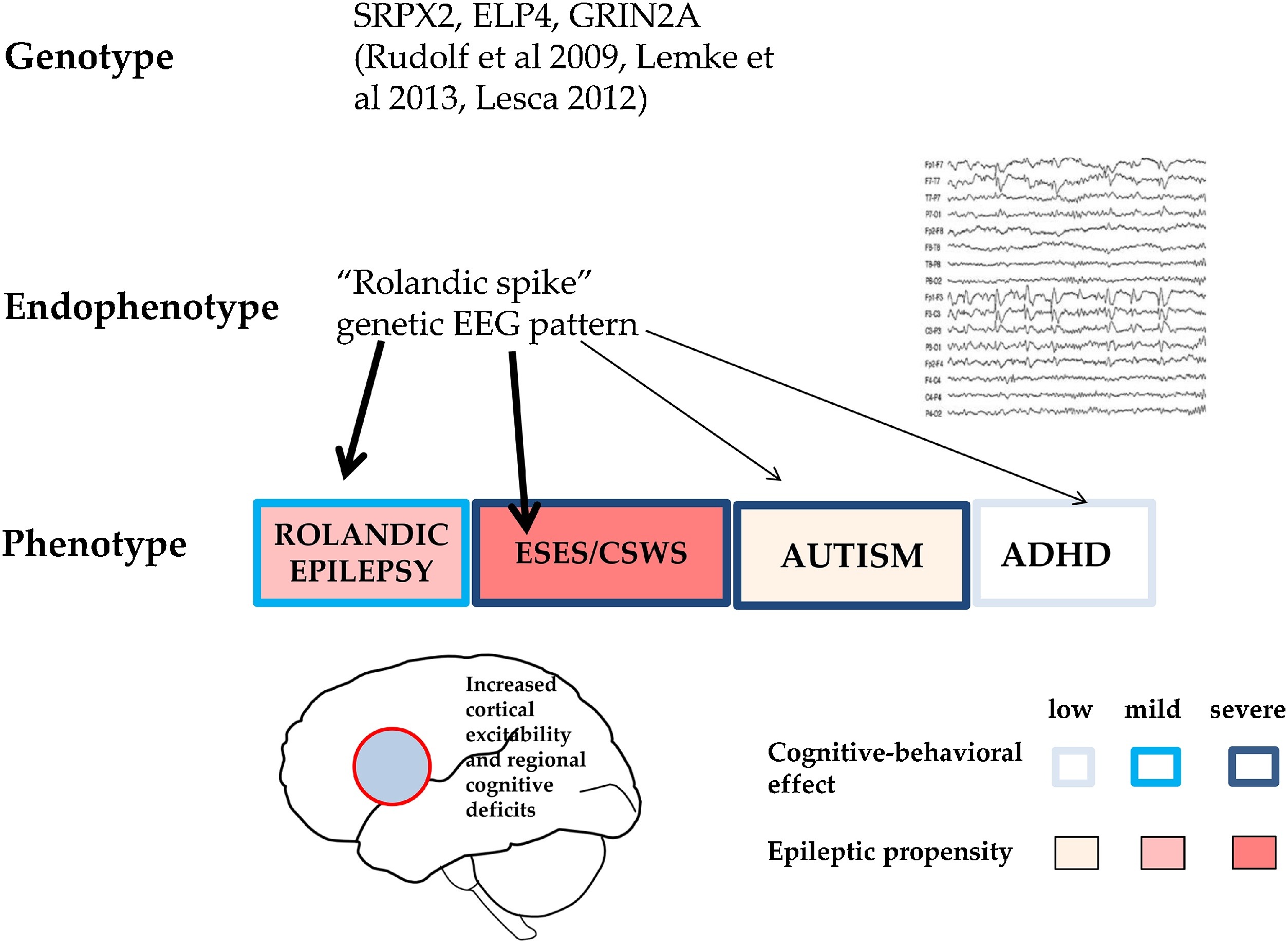
CTS as a shared pattern in ICFE and ESES/LKSs, autism spectrum and ADHD. Schematic representation of epileptic propensity (red) and cognitive impairment (blue). EEG pattern (right). Bottom: The territory of the red circle represents the area of increased epileptic excitability and cognitive-behavioural effects. Related recognised gene-mutations are also shown.
It seems more and more likely that RE, LKS and ESES are different phenotypic variants of the same disease along a spectrum of focal childhood idiopathic epilepsies. The key factor in these epilepsies is the endophenotypic propensity to increase IEDs during sleep, resulting in cognitive loss.
ESES may start in early childhood and cease before puberty. The traditional definition used to require pathologic discharges in 85% of NREM sleep. Now, 25–85% is considered sufficient for the diagnosis (Sánchez Fernández et al., 2012a, 2012b). The severity of cognitive deficit in ESES correlates with its duration and the clinical reversibility depends on its disappearance. The topography of discharges determines the type of cognitive deficit. In cases where RE and PS patients had sleep recordings prior to the development of ESES the sleep-discharges were similar to those seen later in ESES, just quantitative differences in the frequency, voltage, synchrony and spread of IEDs were seen.
We start to understand the way of cognitive decline. Sleep-related memory consolidation makes slow wave sleep the pledge of learning, justifying the need and the seemingly uneconomical presence and persistence of sleep across the phylogeny. Epileptic discharges interweaving NREM sleep may interfere with its plastic functions; vividly depictured by Tassinari in the description of „Penelope syndrome”1 (Tassinari et al., 2009).
Recent studies on the sleep of ESES patients indirectly suggest the deficit of night-time synaptic downscaling (Bölsterli et al., 2011, 2017,Cantalupo et al., 2011) expected on the basis of the synaptic homeostasis theory (Tononi and Cirelli., 2003). Recently, Bölsterli, et al. (2017) have shown that after the recovery from ESES the normal overnight decline of slow waves restored, and the outcome of ESES was better if this overnight decline of sleep slow waves was somewhat preserved during the active ESES period.
Taking together, we have seen three examples how epilepsy changes major brain systems by derailing physiological NREM sleep constituents into epileptic working modes. IEDs seem to exert more system-specific cognitive harm than seizures do
The common step in the first two demonstrated conditions (absence epilepsy and MTLE) is the epileptic transformation of a physiological NREM sleep circuit by the derailment of system-specific plasticity. In absence epilepsy, the epileptic derailment of spindling leads to bilateral SW activity during the burst–firing working mode of the cortico-thalamic system, while in MTLE the epileptic transformation affects the hippocampal SPW-Rs turning to temporal spikes and pathological HFOs. In the spectrum of perisylvian epilepsies, the epileptic transformation is somewhat different. Here an aberrant development of the perisylvian network‘s cortex manifests in the form of CTSs (with or without ripples), that is the first member of the epileptic transformation, occasionally continuing in two subsequent steps. Firstly, idiopathic childhood regional hyperexcitability syndromes may develop and secondly, a progress to regional or diffuse encephalopathies – LKS and ESES – with almost continuous discharges in NREM sleep and severe cognitive loss- may occur. The stepwise stages of epileptic transformation build a spectrum; the phenotypic variations of the same or similar genetic trends (Fig. 7).
Fig. 7.
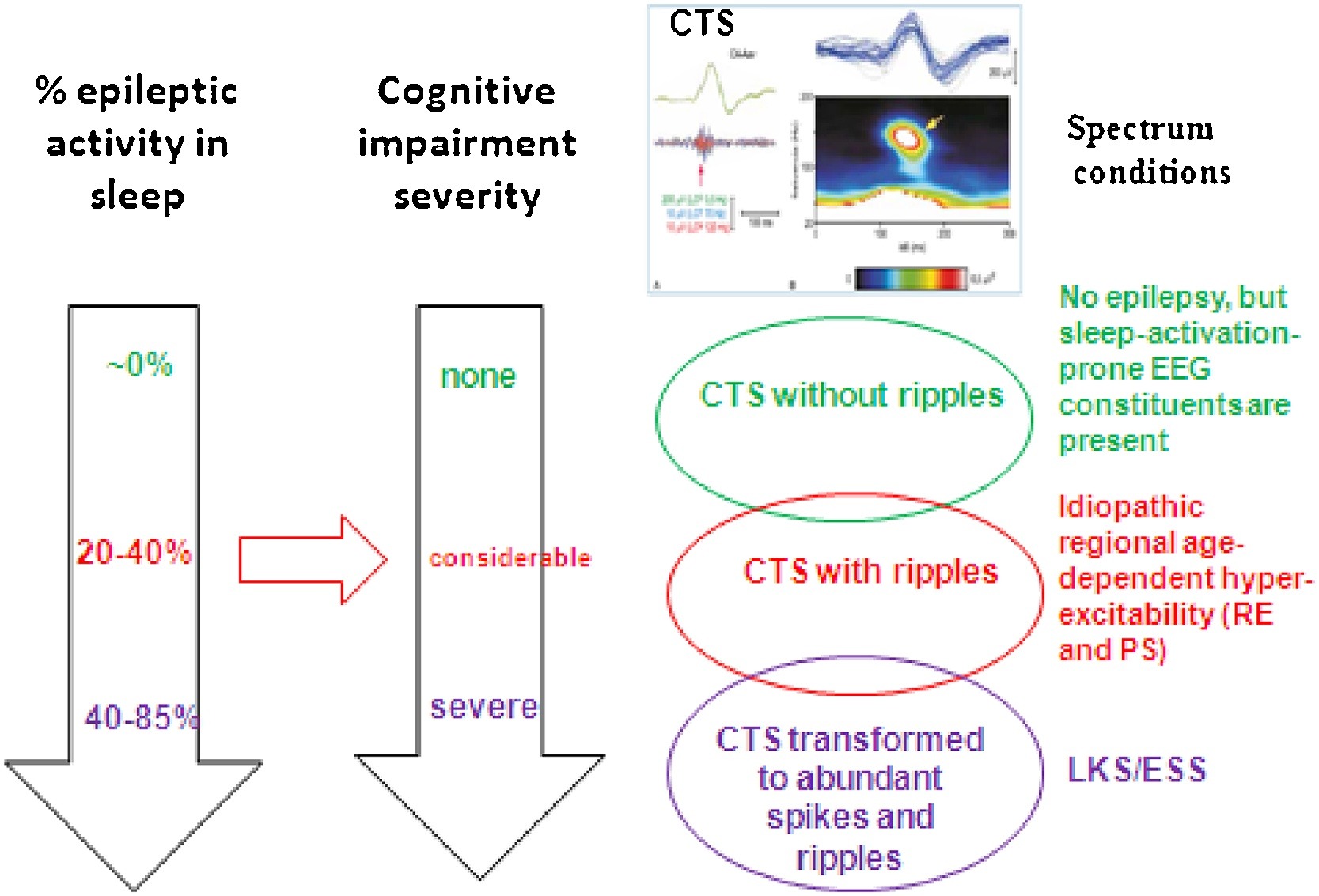
Spectrum disorders of the perisylvian epileptic network. The three rows show the individual constellations. TOP: CTS without ripple and seizures; Middle: Rolandic epilepsy and Panayiotopoulos syndrome with CTS + ripples; Bottom: ESES/LKS with transformation to spikes and HFOs. The perpendicular arrows show the increasing amount of spiking in sleep (estimated data) and the parallel progressive cognitive impairment Red horizontal arrow shows the hypothesized causal relationship between the amount of spikes+HFOs and the degree of the cognitive decline. (C) Centro-temporal spike crowned by a 120 Hz ripple (after the work of Kobayashi et al 2011).
The circuits and oscillations of NREM sleep plastic functions play essential role in these epileptic transformations, creating the strong bond between sleep and epilepsy.
- Discussion
Contemporary sleep research has evidenced that memory consolidation and synaptic recovery mainly occur during NREM sleep providing learning capacities for the subsequent day. This is an important biological function of sleep explaining its subsistence during the phylogeny. There are plenty of examples for the exaggerations and epileptiform transformations of sleep EEG patterns connected with plasticity e.g., in the hippocampus and the thalamo-cortical network (Steriade and Timofejev, 2003; Buzsáki, 2015.). What is the reason for the common affinity of NREM sleep and epilepsy to the circuits and oscillations of learning and cognitive functioning; in other words, why does NREM sleep favor epileptic transformation? We hypothesize that in the group of sleep related epilepsies, the main pathophysiology step is a derailment of sleep-plasticity.
We analyze here two topics: 1) the link of IEDs to sleep oscillations serving plastic functions and 2) the role of NREM sleep plastic functions in the epileptic transformation of certain physiological brain systems.
1 IEDs during NREM sleep associate with the emerging phasic slow wave activity within the frames of CAP in the majority of epilepsies. The IEDs of the perisylvian network spectrum disorders make an exception coupling with sleep spindles (Beelke et al., 2000; Nobili et al., 2001). This interesting dichotomy has remained unexplained; however, it contributes to the delineation of the perisylvian spectrum disorders as a coherent group. Although the important role of slow waves and spindles is evident in memory and learning (Latchoumane et al., 2018), their particular distinctive role is unclear. Nevertheless, the coupling of CTSs with spindles seen in perisylvian spectrum conditions, in a system deeply involved in human communication and cognitive functions, cannot be incidental. Recent studies investigating the association of IEDs and sleep oscillations with intracranial electrodes revealed the link of spikes with the up-down states’ bistability of slow waves (Frauscher et al., 2015a, Újma, et al., 2017) possibly also connecting with plasticity.
2 In epilepsies strongly linked to sleep, the epileptic transformation develops in certain NREM sleep circuits and oscillations, particularly designated to plastic functions. The epileptic occupation of these systems leads to the development of memory-specific (MTLE), or general (perysilvian spectrum conditions) cognitive impairment patterns, while in absence epilepsy the cognitive functions remain intact. It is possible that this difference depends on the degree of the involved system’s participation in cognitive functioning, since absence epilepsy mainly affects those structures involved in the regulation of consciousness and vigilance, while in MTLE and perisylvian epilepsies the systems of declarative memory and human communication are affected. Out of the rest (here not treated) of major sleep related epilepsies, idiopathic, non-lesional frontal lobe epilepsy associated to arousal and characterized by few or no IEDs, does not cause severe cognitive decline (Halász, 2015). Therefore, sleep related epilepsies counting for more than half of all epilepsies do not make a uniform group, concerning cognitive impairment.
The emerging role of IEDs in the development of cognitive decline highlights the unresolved need of a real, causative pharmaco-therapy in epilepsy. Antiepileptic drugs do not suppress IEDs and their anti-seizure effect is merely symptomatic. We are armless against the evolution of epileptic networks and against their interference with plastic sleep functions. Epileptology should urge pharmacological research to open new trends accordingly. Since IEDs exert their devastating effect through sleep slow wave deprivation (Bölsterli et al., 2011, 2017; Cantalupo et al., 2011), those methods restoring slow wave sleep may show new vistas in the therapy of cognitive decline. The initiation of such trends may be witnessed in trials boosting cognitive yield by external acoustic stimulation during NREM sleep (Pellicciari et al., 2009; Ngo et al., 2013; Bellesi et al., 2014).
Disclosure
The authors have no conflict of interest to declare.
Acknowledgement
The authors are grafeful for the National Brain Research Program2017-1.2.1-NKP-2017-00002.
References
Altena, E., Van Der Born, J., Rasch, B., Gais, S., 2006. Sleep to remember. Neuroscientist 12, 410–424.
Avoli, M., 2012. A brief history on the oscillating roles of thalamus and cortex in absence seizures. Epilepsia 53, 779–789.
Baglietto, M.G., Battaglia, F.M., Nobili, L., Tortorelli, S., De Negri, E., Calevo, M.G., Veneselli, E., De Negri, M., 2001. Neuropsychological disorders related to interictal epileptic discharges during sleep in benign epilepsy of childhood with centrotemporal or Rolandic spikes. Dev. Med. Child Neurol. 43 (6), 407–412.
Bal, T., von Krosigk, M., McCormick, D.A., 1995. Role of the ferret perigeniculate nucleus in the generation of syn chronized oscillations in vitro. J. Physiol. 483, 665–685.
Bancaud, J., Talairach, J., Morel, P., Bresson, M., Bonis, A., Geier, S., Hemon, E., Buser, P.G., 1974. Generalized epileptic seizures elicited by electrical stimulation of the frontal lobe in man. Electroencephalogr. Clin. Neurophysiol. 37, 275–282.
Bazil, C.W., 2002. Sleep and epilepsy. Semin. Neurol. 22, 321–327.
Beelke, M., Nobili, L., Baglietto, M.G., De Carli, F., Robert, A., De Negri, E., Ferrillo, F., 2000. Relationship of sigma activity to sleep interictal epileptic discharges: a study in children affected by benign epilepsy with occipital paroxysms. Epilepsy Res. 40, 179–186.
Beenhakker, M.P., Huguenard, J.R., 2009. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron 62, 612–632.
Bellesi, M., Riedner, B.A., Garcia-Molina, G.N., Cirelli, C., Tononi, E., 2014. Enhancement of sleep slow waves: underlying mechanisms and practical consequences. Front. Syst. Neurosci. 28, 208.
Blumenfeld, H., Klein, J.P., Schridde, U., Vestal, M., Rice, T., Khera, D.S., Bashyal, C., Giblin, K., Paul-Laughinghouse, C., Wang, F., Phadke, A., Mission, J., Agarwal, R.K., Englot, D.J., Motelow, J., Nersesy, H., Waxman, S.G., Levin, A.R., 2008. Early treatment supresses the development of spike wave epilepsy in a rat model. Epilepsia 49, 400–409.
Bolsterli, B.K., Schmitt, B., Bast, T., Critelli, H., Heinzle, J., Jenni, O.G., Huber, R., 2011. Impaired slow wave sleep downscaling in encephalopathy with status epilepticus during sleep (ESES). Clin. Neurophysiol. 122, 1779–1787.
Bolsterli, B.K., Gardella, E., Pavlidis, E., Wehrle, F.M., Tassinari, C.A., Huber, R., Rubboli, G., 2017. Remission of encephalopathy with status epilepticus (ESES). During sleep renormalizes regulation of slow wave sleep. Epilepsia 58, 1892–1901.
Boly, M., Jones, B., Findlay, G., Plumley, E., Mensen, A., Hermann, B., Tononi, G., Maganti, R., 2017. Altered sleep homeostasis correlates with cognitive impairment in patients with focal epilepsy. Brain 140, 1026–1040.
Boufidis, S., Rikou, K., Karlovasssiou, A., Vlahoyanni, E., Balyannis, S.I., Kosmoidis, M.H., 2004. Impact of insomnia on working memory. J. Korean Sleep Res. Soc. 13 (Suppl. 1), 91 Proceedings of the European Sleep Medicine Research Society Congress.
Buzsaki, G., 1986. Hippocampal sharp waves: their origin and significance. Brain Res. 398, 242–252.
Buzsaki, G., 1989. Two-stage model of memory trace formation: a role for “noisy” brain states. Neuroscience 31, 551–570.
Buzsaki, G., 2015. Hippocampal Sharp Wave-Ripple: a cognitive biomarker for episodic memory and planning. Hippocampus 25, 1073–1188.
Cajochen, C., Foy, R., Dijk, D.J., 1999. Frontal predominance of a relative increase in sleep delta and theta EEG activity after sleep loss in humans. Sleep Res. Online 2, 65–99.
Cantalupo, G., Rubboli, G., Tassinari, C.A., 2011. Night-time unravelling of the brain web: impaired synaptic downscaling in ESES-the Penelope syndrome. Clin. Neurophysiol. 122, 1691–1692.
Chan, S., Pressler, R., Boyd, S.G., Baldeweg, T., Cross, J.H., 2017. Does sleep benefit memory consolidation in children with focal epilepsy? Epilepsia 58, 456–466.
Cheng, S., Frank, L.M., 2008. New experiences enhance coordinated neural activity in the hippocampus. Neuron 24, 303–313.
Chrobak, J.J., Buzsaki, G., 1998. Operational dynamics in the hippocampal-entorhinal axis. Neurosci. Biobehav. Rev. 22, 303–310.
Clemens, Z., Molle, M., Erőss, L., Jakus, R., Rasonyi, G., Halasz, P., Born, J., 2011. Finetuned coupling between human parahippocampal ripples and sleep spindles. Eur. J. Neuro Sci. 33, 511–520.
Colrain, I.M., Crowley, K.E., Nicholas, C.L., Afifi, L., Baker, F.C., Padilla, M., Turlington, S.R., Trinder, J., 2010. Sleep evoked delta frequency responses show a linear decline in amplitude across the adult lifespan. Neurobiol. Aging 31, 874–883.
Csicsvari, J., Hirase, H., Czurko, A., Mamiya, A., Buzsaki, G., 1999. Oscillatory coupling of hippocampal pyramidal cells and inter neurons in the behaving rat. J. Neurosci. 1, 274–287.
Dalla Bernardina, B., Tassinari, C.A., Dravet, C., Bureau, M., Beghini, G., Roger, J., 1978. Benign focal epilepsy and “electrical status epilepticus” during sleep. Rev. Electroencephalogr. Neurophysiol. Clin. 8, 350–353.
Dalla Bernardina, B., Sgro, V., Caraballo, R., Fontana, E., Colamaria, V., Zullini, E., Simone, M., Zanetti, R., 1991. Sleep and benign partial epilepsies of childhood: EEG and evoked potentials study. Epilepsy Res. Suppl. 2, 83–96.
Danhofer, P., Pejčochova, J., Dušek, L., Rektor, I., Ošlejškova, H., 2018. The influence of EEG-detected nocturnal centrotemporal discharges on the expression of core symptoms of ADHD in children with benign childhood epilepsy with centrotemporal spikes (BCECTS): a prospective study in a tertiary referral center. Epilepsy Behav. 79, 75–81.
de Marco, P., Tassinari, C.A., 1981. Extreme somatosensory evoked potential (ESEP): an EEG sign forecasting the possible occurrence of seizures in children. Epilepsia 22 569-567.
Di Negri, M., 1997. Electrical status epilepticus during sleep (ESES). Different clinical syndromes: towards a unifying view? Brain Dev. 19 (7), 447–451.
Diekelmann, S., Born, J., 2010. The memory function of sleep. Nat. Rev. Neurosci. 11, 114–126.
Dinkelacker, V., Xin, X., Baulac, M., Samson, S., Dupont, S., 2016. Interictal epileptic discharge correlates with global and frontal cognitive dysfunction in temporal lobe epilepsy. Epilepsy Behav. 62, 197–203.
Dinner, D.S., Luders, H.O., 2001. Epilepsy and Sleep. Academic Press, San Diego.
Doose, H., Baier, W.K., 1989. Benign partial epilepsy and related conditions: multifactoral pathogenesis with hereditary impairment of brain maturation. Eur. J. Pediatr. 149, 152–158.
Durmer, J.S., Dinges, D.F., 2005. Neurocognitive consequences of sleep deprivation. Semin. Neurol. 25, 117–129.
Fejerman, N., 2009. Atypical rolandic epilepsy. Epilepsia 50 (Suppl 7), 9–12.
Finelli, L.A., Borbely, A.A., Achermann, P., 2001. Functional topography of the human nonREM sleep electroencephalogram. Eur. J. Neurosci. 13, 2282–2290.
Fogel, S.M., Smith, C.T., 2006. Learning-dependent changes in sleep spindles and Stage 2 sleep. J. Sleep Res. 15, 250–255.
Foldvary-Schaefer, N., Grigg-Damberger, M., 2006. Sleep and epilepsy: what we know, don’t now, and need to know. J. Clin. Neurophysiol. 23, 4–20.
Fonesca, L.C., Tedrus, G.M., 1994. Epileptic syndromes in children wirth somatosensory evoked spikes. Clin. Electroencepalogr. 25, 54–58.
Foster, D.J., Wilson, M.A., 2006. Reverse replay of behavioural sequences in hippocampal place cells during the awake state. Nature 440, 680–683.
Frank, M.G., 2012. Erasing synapses in sleep: is it time to Be SHY? Neural Plast. 264378.
Frauscher, B., Bernasconi, N., Caldairou, B., von Ellenrieder, N., Bernasconi, A., Gotman, J., Dubeau, F., 2015a. Interictal hippocampal spiking influences the occurence of hippocampal. Sleep Spindles. Sleep 38, 1927–1933.
Frauscher, B., von Ellenrieder, N., Ferrari-Marinho, T., Avoli, M., Dubeau, F., Gotman, J., 2015b. Facilitation of epileptic activity during sleep is mediated by high amplitude slow waves. Brain 138 (Pt 6), 1629–1641.
Frauscher, B., Bartolomei, F., Kobayashi, K., Cimbalnik, J., van’ t Klooster, M.A., Rampp, S., Otsubo, H., Holler, Y., Wu, J.Y., Asano, E., Engel Jr., J., Kahane, P., Jacobs, J., Gotman, J., 2017. High-frequency oscillations: the state of clinical research. Epilepsia 58, 1316–1329.
Gais, S., Molle, M., Helms, K., Born, J., 2002. Learning-dependent increases in sleep spindle density. J. Neuro Sci. 22, 6830–6834.
Gelinas, J.N., Khodagholy, D., Thesen, T., Devinsky, O., Buzsaki, G., 2016. Interictal epileptiform discharges induce hippocampal-cortical coupling in temporal lobe epilepsy. Nat. Med. 22, 641–648.
Girardeau, G., Benchenane, K., Wiener, S.I., Buzsaki, G., Zugaro, M.B., 2009. Selective suppression of hippocampal ripples impairs spatial memory. Nat. Neurol Sci. 12, 1222–1223.
Gloor, P., 1968. Generalized cortico-reticular epilepsies. Some considerations on the pathophysiology of generalized bilaterally synchronous spike and wave discharge. Epilepsia 9, 249–263.
Gloor, P., 1979. Generalized epilepsy with spike-and-wave discharge: a reinterpretation of its electrographic and clinical manifestations. The 1977 William G. Lennox Lecture, American Epilepsy Society. Epilepsia 20, 571–588.
Gloor, P., Testa, G., Guberman, A., 1973. Brain-stem and cortical mechanisms in an animal model of corticoreticular epilepsy. Trans. Am. Neurol. Assoc. 98, 203–205.
Goddard, G.V., Douglas, R.M., 1975. Does the engram of kindling model the engram of normal long term memory? Can. J. Neurol. Sci. 2, 385–394.
Grosmark, A.D., Mizuseki, K., Pastalkova, E., Diba, K., Buzsaki, G., 2012. Neuron 75, 1001–1007.
Gulyas, A.I., Freund, T.T., 2015. Generation of physiological and pathological high frequency oscillations: the role of per isomatic inhibition insharp-wave ripple and interictal spike generation. Curr. Opin. Neurobiol. 31, 26–32.
Halasz, P., 1982. Role of the Non-specific Phasic Activation in Sleep Regulation and in the Pathomechanism of Generalized Epilepsy with Spike-wave Pattern. Doctoral thesis, Budapest.
Halasz, P., 2015. Are absence epilepsy and nocturnal frontal lobe epilepsy system epilepsies of the sleep/wake system? Behav. Neurol. 231676.
Halasz, P., Kelemen, A., Clemens, B., Saracz, J., Rosdy, B., Rasonyi, G., Szűcs, A., 2005. The perisylvian epileptic network. A unifying concept. Ideggyogy. Sz. 58, 21–31.
Hauri, P.J., 1997. Cognitive deficits in insomnia patients. Acta Neurolgica Belgica 97, 177–184.
Horita, H., Uchida, E., Maekawa, K., 1991. Circadian rhythm of regular spike-wave discharges in childhood absence epilepsy. Brain Dev. 13, 200–202.
Horne, J.A., 1993. Human sleep, sleep loss and behaviour: implications for the prefrontal cortex and psychiatric disorder. Br. J. Psychiatry 162, 413–419.
Huber, R., Ghilardi, M., Massimini, M., Ferrarelli, F., Riedner, B.A., Peterson, M.J., Tononi, G., 2006. Arm immobilization causes cortical plastic changes and locally decreases sleep slow wave activity. Nat. Neurosci. 9, 1169–1176.
Huguenard, J.R., McCormick, D.A., 2007. Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci. 30 (7), 350–356.
Huguenard, J.R., Prince, D.A., 1994. Clonazepam suppresses GABAB-mediated inhibition in thalamic relay neurons through effects in nucleus reticularis. J. Neurophysiol. 71, 2576–2581.
Jasper, H.H., 1977. Wilder Penfield: his legacy to neurology. The centrencephalic system. Can. Med. Assoc. J. 116 (12), 1371–1372.
Jasper, H.H., Droogleever-Fortuyn, J., 1946. Experimental studies of the functional anatomy of petit mal epilepsy. Res. Publ. Ass. Res. Nerv. Ment. Dis. 26, 272–298.
Ji, D., Wilson, M.A., 2007. Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat. Neurosci. 10, 100–107.
Kattler, H., Dijk, D.J., Borbely, A.A., 1994. Effect of unilater al somatosensory stimulation prior to sleep on the slee p EEG in humans. J. Sleep Res. 3, 159–164.
Kellermann, K., 1978. Recurrent aphasia with subclinical bioelectric status epilepticus during sleep. Eur. J. Pediatr. 3 128 (3), 207–212.
Kleen, J., Scott, R.C., Holmes, G.L., Roberts, D.W., Rundle, M.M., Testorf, M., Lenck-Santini, P.P., Jobst, B.C., 2013. Hippocampal interictal epileptiform activity disrupts cognition in humans. Neurology 81, 18–24.
Klein, J.P., Khera, D.S., Nersesyan, H., Kimchi, E.Y., Waxman, S.G., Blumenfeld, H., 2004. Dysregulation of sodium channel expression in cortical neurons in a rodent model of absence epilepsy. Brain Res. 1000, 102–109.
Kostopoulos, G.K., 2000. Spike-and-wave discharges of absence seizures as a transformation of sleep spindles: the continuing development of a hypothesis. Clin. Neurol. Physiol. 111 (Suppl 2), S27–38.
Kostopoulos, G.K., 2001. Involvement of the thalamo-cortical system in epileptic loss of consciousness. Epilepsia 42 (Suppl 3), 13–19.
Koutroumanidis, M., Panayiotopoulos, C.P., 1993. Benign childhood partial epilepsies: benign childhood seizure susceptibility syndromes. J. Neurol. Neurosurg. Psychiatr. 56, 2–5.
Latchoumane, C.V., Ngo, H.V., Born, J., Shin, H.S., 2018. Thalamic spindles promote memory formation during sleep through triple phase-locking of cortical, thalamic, and hippocampal rhythms. Neuron 95, 424–435.
Lemke, J.R., Lal, D., Reinthaler, E.M., Steiner, I., Nothnagel, M., Alber, M., Geider, K., Laube, B., et al., 2013. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 45, 1067–1072.
Lesca, G., Rudolf, G., Labalme, A., Hirsch, E., Arzimanoglou, A., Genton, P., Motte, J., de Saint Martin, A., Valenti, M.P., Boulay, C., De Bellescize, J., Keo-Kosal, P., Boutry-Kryza, N., Edery, P., Sanlaville, D., Szepetowski, P., 2012. Epileptic encephalopathies of the Landau-Kleffner and continuous spike and waves during slow-wave sleep types: genomic dissection makes the link with autism. Epilepsia 53, 1526–1538.
Lewis, P.A., Durrant, S.J., 2011. Overlapping memory replay during sleep builds cognitive schemata. Trends Cogn. Sci. 15, 343–351.
Manning, J.P., Richards, D.A., Leresche, N., Crunelli, V., Bowery, N.G., 2004. Cortical-area specific block of genetically determined absence seizures by ethosuximid. Neuroscience 123, 5–9.
Meeren, H., Pijn, J.P., Van Luijtelaar, E.L., Coenen, A.M., Lopes da Silva, F.H., 2002. Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J. Neurosci. 22, 1480–1495.
Meeren, H., van Luijtelaar, G., Lopes da Silva, F., Coenen, A., 2005. Evolving concepts on the pathophysiology of absence seizures: the cortical focus theory. Arch. Neurol. 62, 371–376.
Mendez, M., Radtke, R.A., 2001. Interactions between sleep and epilepsy. J. Clin. Neurophysiol. 18, 106–127.
Milner, B., 2005. The medial temporal-lobe amnesic syndrome. Psychiatr. Clin. North Am. 28, 599–611.
Molle, M., Born, J., 2011. Slow oscillations orchestrating fast oscillations and memory consolidation. Prog. Brain Res. 193, 93–110.
Molle, M., Bergmann, T.O., Marshall, L., Born, J., 2011. Fast and slow spindles during the sleep slow oscillation: disparate coalescence and engagement in memory processing. Sleep 34, 1411–1421.
Nadasdy, Z., 2000. Spike sequences and their consequences. J. Physiol. Paris 94, 505–524.
Nass, R., Devinsky, O., 1999. Autistic regression with rolandic spikes. Neuropsychiatry Neuropsychol. Behav. Neurol. 12, 193–197.
Neumann, H., Helmke, F., Thiels, C., Polster, T., Selzer, L.M., Daseking, M., Petermann, F., Lucke, T., 2016. [Cognitive development in children with benign rolandic epilepsy of childhood with centrotemporal spikes – results of a current systematic database search]. Fortschr. Neurol. Psychiatr. 84, 617–632.
Ngo, H.V., Claussen, J.C., Born, J., Molle, M., 2013. Induction of slow oscillations by rhythmic acoustic stimulation. J. Sleep Res. 22, 22–31.
Niedermeyer, E., 1972. The Generalized Epilepsies. Thomas, Springfield. Niedermeyer, E., Laws, E.R., Walker, E.A., 1969. Depth EEG findings in epileptic patients with generalized spike -wave complexes. Arch. Neurol. 21, 51–58.
Niethard, N., Burgalossi, A., Born, J., 2017. Plasticity during sleep is linked to specific regulation of cortical circuit activity. Front. Neural Circuits 11, 65.
Nobili, L., Ferrillo, F., Baglietto, M.G., Beelke, M., De Carli, F., De Negri, E., et al., 1999. Relationship of sleep interictal epileptiform discharges to sigma activity (12-16 Hz) in benign epilepsy of childhood with Rolandic spikes. Clin. Neurophysiol. 110, 39–46.
Nobili, L., Baglietto, M.G., Beelke, M., De Carli, F., De Negri, E., Gaggero, R., Rosadini, G., Veneselli, E., Ferrillo, F.D., 2001. Distribution of epileptiform discharges during nREM sleep in the CSWSS syndrome: relationship with sigma and delta activities. Epilepsy Res. 44, 119–128.
O’Neil, J., Senior, T.J., Allen, K., Huxter, J.R., Csicsvari, J., 2000. Reactivation of experience-dependent cell assembly patterns in the hippocampus. Nat. Neurosci. 11, 209–215.
Pal, D.K., Ferrie, C., Addis, L., Akiyama, T., Capovilla, G., et al., 2016. Idiopathic focal epilepsies: the “lost tribe”. Epileptic Disord. 18, 252–288.
Parrino, L., Smerieri, A., Spaggiari, M.C., Terzano, M.G., 2000. Cyclic alternating pattern (CAP) and epilepsy during sleep: how a physiological rhythm modulates a pathological event. Clin. Neurophysiol. 111 (Suppl 2), S39–46.
Passouant, P., Besset, A., Carriere, A., Billiard, M., 1974. In: Koella, W.P., Levien, P. (Eds.), Night Sleep and Generalized Epilepsy, Sleep. Karger, Basel, pp. 185–196.
Patry, G., Lyagoubi, S., Tassinari, C.A., 1971. Subclinical electrical status epilepticus induced by sleep in children. Arch. Neurol. 24, 242–252.
Payne, J.D., Kensinger, E.A., 2011. Sleep leads to changes in the emotional memory trace: evidence from FMRI. J. Cogn. Neurosci. 23, 1285–1297.
Pellicciari, M.C., Miniussi, C., Rossini, P.M., De Gennaro, L., 2009. Increased cortical plasticity in the elderly: changes in the somatosensory cortex after paired associative stimulation. Neuroscience 63, 266–276.
Penfield, W., Jasper, H.H., Penfield, W., 1954. Epilepsy and the Functional Anatomy of the Human Brain. Little, Brown, Boston.
Potari, A., Ujma, P.P., Konrad, B.N., Genzel, L., Simor, P., Kormendi, J., Gombos, F., Steiger, A., Dresler, M., Bodizs, R., 2017. Age-related changes in sleep EEG are attenuated in highly intelligent individuals. Neuroimage 146, 554–560.
Puertas, F.J., Tembl, A., Cordero, J.M., Azzi, H., Romero, M.F., Merino, A., 2004. Changes in cerebral perfudion studied by SPECT in patients with severe obstructive apnea/hypopneasyndrome J. Sleep Res. 592 Proceedings of the 24th European Sleep Research Society Congress.
Rajna, P., Lona, C., 1989. Sensory stimulation for inhibition of epileptic seizures. Epilepsia 30, 168–174.
Rasch, B., Born, J., 2013. About sleep’s role in memory. Physiol. Rev. 93, 681–766.
Rossi, P.G., Parmeggiani, A., Bach, V., Santucci, M., Visconti, P., 1995. EEG features and epilepsy in patients with autism. Brain Dev. 17, 169–174.
Roux, L., Hu, B., Eichler, R., Stark, E., Buzsaki, G., 2017. Sharp wave ripples during learning stabilize the hippocampa Coordinated memory replay in the visual cortex and hippocampus during sleep. Nat. Neurosci. 20, 845–853.
Rudolf, G., Valenti, M.P., Hirsch, E., Szepetowski, P., 2009. From rolandic epilepsy to continuous spike-and-waves during sleep and Landau-Kleffner syndromes: insights into possible genetic factors. Epilepsia 50 (Suppl 7), 25–28.
Saltik, S., Uluduz, D., Cokar, O., Demirbilek, V., Dervent, A., 2005. A clinical and EEG study on idiopathic partial epilepsies with evolution into ESES spectrum disorders. Epilepsia 46, 524–533.
Sanchez Fernandez, I., Loddenkemper, T., Peters, J.M., Kothare, S.V., 2012a. Electrical status epilepticus in sleep: clinical presentation and pathophysiology. Pediatr. Neurol. 47, 390–410.
Sanchez Fernandez, I., Peters, J.M., Hadjiloizou, S., Prabhu, S.P., Zarowski, M., Stannard, K.M., Takeoka, M., Rotenberg, A., Kothare, S.V., Loddenkemper, T., 2012b. Clinical staging and electroencephalographic evolution of continuous spikes and waves during sleep. Epilepsia 53, 1185–1195.
Sanchez-Vives, M.V., Bal, T., McCormick, D.A., 1995. Properties of GABAergic inhibition in the ferret LGNd contributing to the generation of synchronized oscillations. Soc. Neurosci. Abst. 21, 11.
Schabus, M., Gruber, G., Parapatics, S., Sauter, C., Klosch, G., Anderer, P., Klimesch, W., Saletu, B., Zeitlhofer, J., 2004. Sleep spindles and their significance for declarative memory consolidation. Sleep 27, 1479–1485.
Schlingloff, D., Kali, S., Freund, T.F., Hajos, N., Gulyas, A.I., 2014. Mechanisms of sharp wave initiation and ripple generation. J. Neurosci. 34, 11385–11398.
Shatskikh, T.N., Raghavendra, M., Zhao, Q., Cui, Z., Holmes, G.L., 2006. Electrical induction of spikes in the hippocampus impairs recognition capacity and spatial memory in rats. Epilepsy Behav. 9, 549–556.
Siapas, A.G., Wilson, M.A., 1998. Coordinated interactions spindles during slow-wave sleep. Neuron 21, 1123–1128.
Silvestri, R., Gagliano, A., Calarese, T., Arico, I., Cedro, C., Condurso, R., Germano, E., Vita, G., Tortorella, G., 2007. Ictal and interictal EEG abnormalities in ADHD children recorded over night by video-polysomnography. Epilepsy Res. 75, 130–137.
Staresina, B.P., Bergmann, T.O., Bonnefond, M., van der Meij, R., Jensen, O., Deuker, L., Elger, C.E., Axmacher, N., Fell, J., 2015. Hierarchical nesting of slow oscillations, spindles and ripples in the human hippocampus during sleep. Nat. Neuro. Sci. 18, 1679–1686.
Steriade, M., 2003a. Neuronal Substrates of Sleep and Epilepsy. Cambridge University Press, pp. 67–73.
Steriade, M., 2003b. Neuronal Substrates of Sleep and Epilepsy. Cambridge University Press, pp. 132.
Steriade, M., Timofejev, I., 2003. Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron 37 (563), -576.
Steriade, M., Deschenes, M., Domich, L., Mulle, C., 1985. Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J. Neurophysiol. 54, 1473–1497.
Stevens, J.R., Kodama, H., Lonsbury, B., Mills, L., 1971. Ultradian characteristics of spontaneous seizure discharges recorded by radio telemetry in man. Electroencephalogr. Clin. Neurophysiol. 31, 313–325.
Tamminen, J., Payne, J.D., Stickgold, R., Wamsley, E.J., Gaskell, M.G., 2010. Sleep spindle activity is associated with the integration of new memories and existing knowledge. J. Neurosci. 30, 14356–14360.
Tassinari, C.A., Dravet, C., Roger, J., 1977. Encephalopathy related to electrical status epilepticus during slow sleep. Electroencephal. Clin. Neurophysiol. 43, 529–530.
Tassinari, C.A., Rubboli, G., Volpi, L., Meletti, S., d’Orsi, G., Franca, M., Sabetta, A.R., Riguzzi, P., Gardella, E., Zaniboni, A., Michelucci, R., 2000. Encephalopathy with electrical status epilepticus during slow sleep or ESES syndrome including the acquired aphasia. Clin. Neurophysiol. 111 (Suppl 2), S94–S102.
Tassinari, C.A., Cantalupo, G., Rios-Pohl, L., Giustina, E.D., Rubboli, G., 2009.
Encephalopathy with status epilepticus during slow sleep: “the Penelope syndrome”. Epilepsia 50 (Suppl 7), 4–8.
Terzano, M.G., Parrino, L., Smerieri, A., Carli, F., Nobili, L., Donadio, S., Ferrillo, F., 2005.
CAP and arousals are involved in the homeostatic and ultradian sleep processes. J. Sleep Res. 14, 359–368.
Tononi, G., Cirelli, C., 2003. Sleep and synaptic homeostasis: a hypothesis. Brain Res. Bull. 62, 143–150.
Tononi, G., Cirelli, C., 2006. Sleep function and synaptic homeostasis. Sleep Med. Rev. 10. 49-46.
Ujma, P.P., Simor, P., Ferri, R., Fabo, D., Kelemen, A., Erőss, L., Bodizs, R., Halasz, P., 2014. Increased interictal spike activity associated with transient slow wave trains during non-rapid eye movement sleep. Sleep Biol. Rhythms 13, 155–162.
Ujma, P.P., Halasz, P., Kelemen, A., Fabo, D., Erőss, L., 2017. Epileptic interictal discharges are more frequent during NREM slow wave downstates. Neurosci. Lett. 658, 37–42.
Ulrich, D., 2016. Sleep spindles as facilitators of memory formation and learning. Neural Plast., 1796715.
Van Bogaert, P., Urbain, C., Galer, S., Ligot, N., Peigneux, P., De Tiege, X., 2012. Impact of focal interictal epileptiform discharges on behaviour and cognition in children. Neurophysiol. Clin. 42 (1-2), 53–58.
Vannest, J., Tenney, J.R., Gelineau-Morel, R., Maloney, Glauser, T.A., 2015. Cognitive and behavioral outcomes in benign childhood epilepsy with centrotemporal spikes. Epilepsy Behav. 45, 85–91.
Vyazovskiy, V., Borbely, A.A., Tobler, I., 2000. Unilateral vibrissae stimulat ion during waking induces interhemispheric EEG asymmetry during subsequent sleep in the rat. J. Sleep Res. 9, 367–371.
Wickens, S., Bowden, S.C., D’Souza, W., 2017. Cognitive functioning in children with selflimited epilepsy with centrotemporal spikes: a systematic review and meta-analysis. Epilepsia 58, 1673–1685.
Williams, D., 1965. The thalamus and epilepsy. Brain 88, 539–556.
Zijlmans, M., Jiruska, P., Zelmann, R., Leijten, F.S., Jefferys, J.G., Gotman, J., 2012. Highfrequency oscillations as a new biomarker in epilepsy. Ann. Neurol. 71, 169–178.
Copyright ©2019. Elsevier Inc.