Research in Developmental Disabilities 35 (2014) 3226 – 3235
DOI: 10.1016/j.ridd.2014.07.056
Róbert Bódizsa,b,*, Ferenc Gombosb, Patrícia Gervánb, Katalin Szőcsc, János M. Réthelyic, Ilona Kovácsb, a
a Institute of Behavioural Sciences, Semmelweis University, Nagyvárad tér 4, H-1089 Budapest, Hungary
b Department of General Psychology, Pázmány Péter Catholic University, Mikszáth tér 1, H-1088 Budapest, Hungary
c Department of Psychiatry and Psychotherapy, Semmelweis University, Balassa u. 6, H-1083 Budapest, Hungary
* Corresponding author at: Institute of Behavioural Sciences, Semmelweis University, Nagyvárad tér 4, H-1089 Budapest, Hungary. Tel.: +36 1 210 2953×56404; fax: +36 1 210 2955. E-mail addresses: bodizs.robert@med.semmelweis-univ.hu (R. Bódizs), fgombos@freemail.hu (F. Gombos), pgervan@gmail.com (P. Gerván), rethelyi.janos@med.semmelweis-univ.hu (J.M. Réthelyi), dr.ilona.kovacs@gmail.com (I. Kovács).
Abstract
Specific developmental and aging trajectories characterize sleep electroencephalogram (EEG) of typically developing (TD) subjects. Williams syndrome (WS) is marked by sleep alterations and accelerated aging of several anatomo-functional and cognitive measures. Here we test the hypothesis of a premature aging of sleep in WS. Age-related changes of home recorded sleep EEG of 42 subjects (21 WS, 21 age- and gender matched TD subjects, age: 6–29 years) were tested by Pearson correlations and homogeneity-of-slopes analysis. Typical developmental/aging effects of sleep EEGs were observed in TD subjects. Accelerated aging in WS was confirmed by overall sleep/wake measures. Specifically, premature aging was evident in accelerated age-dependent declines in WS subjects’ sleep efficiency, as well as in steeper age-related rises in wakefulness and wake after sleep onset (WASO) of the WS group. In contrast, NREM sleep-related measures indicated atypical decelerations of the developmental trends of WS subjects, characterized by the slowing down of the age-related slow wave sleep (SWS) declines mirrored by the lack of agedependent increase in Stage 2 (S2) sleep. Age-effects in sleep EEG power spectra were not different among the groups. Objectively measured sleep disruption of subjects with WS is age-dependent and increasing with age. Moreover, these data suggest atypical pre- and postpubertal neural development in WS, with sleep/wake balance and REM sleep time indicating accelerated aging while NREM sleep composition revealing signs of an as yet unidentified, perhaps compensatory developmental delay.
Keywords: Williams–Beuren syndrome microdeletion, Polysomnography, Sleep EEG, Spectral analysis, Brain development, Aging
Introduction
Williams syndrome (WS, also known as Williams–Beuren syndrome) is a genetically determined neurodevelopmental disorder occurring in 1 of 20,000 live births. The syndrome is caused by a hemideletion of 25–28 genes at 7q11.23 and characterized by mild to moderate mental retardation, learning difficulties, cardiovascular abnormalities, high sociability and empathy and a distinctive cognitive-linguistic profile (Ja¨rvinen-Pasley et al., 2008; Meyer-Lindenberg, Mervis, &Berman, 2006). Attention deficit/hyperactivity disorder (ADHD), specific phobias and generalized anxiety disorder (GAD) are among the particularly frequent comorbid psychiatric syndromes of subjects with WS. The prevalence of GAD in WS was shown to increase significantly with age (Dykens, 2003; Leyfer, Woodruff-Borden, Klein-Tasman, Fricke, & Mervis, 2006). WS is characterized by atypical development of several anatomical measures as well as physiological and cognitive functions, including neurocognitive aspects of language and social communication (Haas & Reiss, 2012; Laing, Hulme, Grant, & Karmiloff-Smith, 2001), visuospatial processes (Atkinson et al., 2001) and motor performance (Tsai, Wu, Liou, & Shu, 2008). Although pubertal development follows a typical pattern, WS girls reach puberty roughly two years earlier than typically developing (TD) girls (Partsch et al., 1999). The early onset puberty was hypothesized to be linked to premature activation of the hypothalamic–pituary axis (Partsch et al., 1999), although the cause of this precocious activation is not identified yet. Reported findings on multiple organ investigations as well as psychological functions are suggestive for the occurrence of mild accelerated aging of subjects with WS (Cherniske, Sadler, Schwartz, Carpenter, & Pober, 1999; Devenny et al., 2004; Krinsky-McHale, Kittler, Brown, Jenkins, & Devenny, 2005). The list of potential indicators includes graying of hair during adolescence or young adulthood (Lenoff, Wang, Greenberg, & Bellugi, 1997; Pober, 2010), cataracts (Cherniske et al., 2004), senile emphysema (Wan, Pober, Washko, Raby, & Silverman, 2010), high frequency sensorineural hearing loss (Marler, Elfenbein, Ryals, Urban, & Netzloff, 2005), premature wrinkling of the skin (Morris, Demsey, Leonard, Dilts, & Blackburn, 1988; Mari et al., 1995), precipitous age-associated decrease in long-term, episodic memory (Devenny et al., 2004), as well as age related semantic memory performance decline (Krinsky-McHale et al., 2005). However, no evidence for age-related decline in social or adaptive functioning in adults with WS was found by others, at least up to the age of 50–55 years (Elison, Stinton, & Howlin, 2010). Similarly, studying inhibitory processing in older patients with WS Greer and colleagues (Greer, Riby, Hamilitona, & Riby, 2013) found no support for accelerated aging hypothesis. Sleep of subjects with WS was shown to be altered in several respects. Difficulties in initiating and maintaining sleep, decreases in total sleep, rapid eye movement (REM) sleep and sleep efficiency, as well as increases in intra-sleep wakefulness, slow wave sleep (SWS) and daytime sleepiness are among the common findings (Annaz, Hill, Ashworth, Holley, & Karmiloff-Smith, 2011; Arens et al., 1998; Ashworth, Hill, Karmiloff-Smith, & Dimitriou, 2013; Bódizs, Gombos, & Kovács, 2012; Goldman, Malow, Newman, Roof, & Dykens, 2009; Gombos, Bódizs, & Kovács, 2011; Mason et al., 2011). Sleep cycles were shown to be fragmented and the cyclicity of sleep disorganized (Gombos et al., 2011). Moreover, increases in non-REM (NREM) sleep frontal electroencephalogram (EEG) delta activity, region-independent decreases in alpha and sigma waves, as well as the accelerations of sigma peak frequencies were also reported (Gombos et al., 2011; Bódizs et al., 2012, 2014). These sleep macrostructural and EEG alterations were shown to be present in children and young adults with WS. Most of the reported alterations in sleep are known to be changing during ontogeny and/or affected by physiological aging in TD subjects. Age is a major determinant of sleep. Almost all of the known sleep architectural or quantitative EEG measures are strongly and reliably depending on the chronological age of the subjects (Carrier, Land, Buysse, Kupfer, & Monk, 2001; Ohayon, Carskadon, Guilleminault, & Vitiello, 2004). Reports on both developmental and aging-related changes in human sleep emphasize age-related decreases in the daily amount of sleep, increases in Stage 2 (S2) sleep percentage, and decreases in SWS percentage (Coble, Kupfer, Taska, & Kane, 1984; Colrain & Baker, 2011; Espiritu, 2008; Iglowstein, Jenni, Molinari, & Largo, 2003; Ohayon et al., 2004). In addition the aging of sleep is characterized by decreases in REM sleep time and increases in wakefulness (Coble et al., 1984; Colrain & Baker, 2011; Espiritu, 2008; Iglowstein et al., 2003; Ohayon et al., 2004). The above changes are reflected in age-related decreases in quantitative EEG measures of sleep EEG delta and theta waves during both NREM and REM sleep (Astro¨m & Trojaborg, 1992; Carrier et al., 2001; Darchia, Campbell, Tan, & Feinberg, 2007; Feinberg & Campbell, 2013; Landolt & Borbély, 2001; Ringli & Huber, 2011). In addition age-related declines in sleep spindling (Nicolas, Petit, Rompré, & Montplaisir, 2001) and/or NREM sleep EEG sigma power (Landolt & Borbély, 2001), as well as the increases in the sigma spectral peak frequency (Tarokh, Carskadon, & Achermann, 2011) and/or in the frequency of sleep spindles was supported by several studies (Crowley, Trinder, Kim, Carrington, & Colrain, 2002; Feinberg, Koresko, & Heller, 1967; Nicolas et al., 2001; Principe & Smith, 1982). In order to shed light on the specific nature of sleep alterations and atypical development in WS as well as to provide further support or disproof of the presumed concept of accelerated aging in WS we report age-related effects in various sleep EEG measures. We hypothesize that the sleep EEG measures which are age-dependently decreasing and increasing in TD subjects are characterized by accelerated age-related decreases and increases in WS subjects, respectively.
Methods
Subjects and genetic investigations
WS participants (N = 21, 7 males and 14 females, age range 6–29 years, mean age standard deviation: 19.19 7.13 years) were contacted through the Hungarian Williams Syndrome Association (parents were mediating in the case of underage subjects). All WS subjects (including adults) were living with their parents. TD controls (N = 21, 6 males and 15 females, age range 6–29 years, mean age standard deviation: 19.14 7.14 years) were selected by personal contacts of the authors and matched by age and sex to the WS participants. A twin pair discordant for WS and sex (Bódizs et al., 2014) was considered as a pair case control.
The clinical diagnosis of WS was established prior to this study by fluorescent in situ hybridization (FISH) test demonstrating the hemideletion of the elastin gene. To confirm the clinical diagnosis and specify the size of the hemideleted region, we carried out multiplex ligation-dependent probe amplification (MLPA) using the SALSA MLPA KIT P029-A1 (MRCHolland, Amsterdam, The Netherlands). Briefly, pairs of locus-specific oligonucleotide probes are hybridized to the target DNA, followed by a ligation and amplification step. Amplified fragments are separated and analyzed using capillary electrophoresis. At the resolution of this genotyping method, all WS subjects were carriers of a typical deletion spanning at least 1.038 Mb between FKBP6 (7:72742167–72772634) and CLIP2 (7:73703805–73820273) (Bódizs et al., 2014). Exclusion criteria for TD participants were medical diagnosis of sleep problems or psychiatric, neurological or other medical disorder. As WS is a rare disease our strategy was to include as many WS subjects as possible, but to document any specificity. Participants were free of drugs except for 1 WS patient who was on stable medication with clonidine (150 mg/ day), enalapril (5 mg/day), acetylsalicylic acid (500 mg/day), betaxololi hydrochloridum (20 mg/day), and amlodipine (15 mg/day). Those patients not on medication were not withdrawn from any pharmacological treatment prior to the study. Adult participants or the parents of the underage participants signed informed consent for the participation in the study according to the Declaration of Helsinki.
Procedures
Subjects’ sleep was recorded at their homes by using ambulatory home polysomnography. Sleep recordings on two consecutive weekend nights were performed according to the subjects’ sleeping habits. We used a portable 32 channel SD LTM Headbox together with a BRAIN QUICK System PLUS software (Micromed, Italy) for polysomnographic data recording. We recorded EEG according to the 10–20 system (Jasper, 1958) at 21 recording sites (Fp1, Fp2, Fpz, F3, F4, F7, F8, Fz, C3, C4, Cz, P3, P4, Pz, T3, T4, T5, T6, O1, O2, Oz) referred to the mathematically linked mastoids. Bipolar EOG, ECG and submental as well as tibialis EMG were also recorded. We have not recorded respiratory variables because there was no indication for any significant breathing difficulty during sleep in the study by Arens et al. (1998), and in a more recent study by Mason et al. (2011). Moreover, this would have caused further inconveniences for the participants because of the recording instruments. However, one participant, whose sleep was one of the most fragmented in our study, underwent a full-night polysomnography in a clinical sleep laboratory before our examination. The results of clinical respiratory monitoring did not show sleep apnea or hypopnea in this participant. EEG and polygraphic data were high-pass filtered at 0.15 Hz and low-pass filtered at 1500 Hz (both 40 dB/decade). Data were collected with an analog to digital conversion rate of 4096 Hz/ channel (synchronous, 16 bit). A further 40 dB/decade anti-aliasing digital filter was applied by digital signal processing (firmware) which low pass filtered the data at 124 Hz. Subsequently, the digitized and filtered EEG was undersampled and stored at a sampling rate of 1024 Hz.
EEG analyses
Sleep recordings of the second nights were visually scored according to standard criteria (Rechtschaffen & Kales, 1968) in 20 second epochs. The following definitions were used for sleep architecture evaluation: total sleep time, sleep efficiency (calculated as the percent of sleep duration/time in bed from lights out), sleep latency (from lights out to first non-Stage 1 [S1] sleep), wake time after sleep onset (WASO as measured from the beginning of first non-S1 sleep), S1 duration, Stage 2 (S2) duration, SWS (defined as the amount of time in Stages 3 [S3] and 4 [S4] sleep) duration, REM sleep duration and REM latency. The next step was the exclusion of artifacts based on the visual inspection of the records. The 4 second epochs containing artifactual sleep EEG (movement, sweating or technical artifacts) were manually removed before further automatic sleep EEG analyses. Average power spectral densities were calculated by a Fast Fourier Transformation (FFT) algorithm applied to the 50 percent overlapping, Hanning-tapered, artifact-free 4 second (4096 points) epochs of whole night non-S1 NREM sleep and REM sleep separately for derivation Fz. The band power values were expressed in mV2s and defined as follows: slow wave/delta activity (0.5–4.5 Hz),1 theta activity (4.75–7.25 Hz), alpha activity (7.5–10.75 Hz), sigma activity (11–15 Hz), beta activity (15.25–30 Hz), and gamma activity (30.25–45 Hz). In order to normalize the distributions, the band power values of both NREM and REM sleep were log-transformed (10th base) before statistical analyses. Logtransformation of spectral values is important from the point of view of the presumed linear age-dependency of sleep EEG power. It was evidenced that the age vs. log-transformed sleep EEG (delta) power relationship is reliably linear in contrast to age vs. raw (non-log-transformed) sleep EEG (delta) power, which is exponential (Aström & Trojaborg, 1992). A spectral peak detection procedure focusing on the 8–16 Hz band of the average power spectra of whole night NREM sleep was performed as follows: the zero-crossing points of the first order derivatives of the spectra were considered as locations of spectral peaks if the second order derivatives were negative at these frequencies (local maxima in mathematical terms). Spectral peak processing was performed on the averaged z-scores of the left and right frontal (F3 and F4), as well as on the left and right parietal (P3 and P4) derivations. Slower and faster sigma peaks are usually dominant over the frontal and
1 A distinction between slow waves below or above 1 Hz (slow oscillation and delta waves) was ignored in our work because of the lack of a difference between the results obtained with these two measures. Thus, the term delta denotes the classical, broad band 0.5–4.5 Hz power in the following parts of the paper.
parietal derivations, respectively. Thus we considered a spectral peak as a slow frontal sigma peak if its frequency was the lowest among the frontally detected peaks (usually one or two peaks were detected). The reverse was implemented for the definition of fast parietal sigma peaks: these were defined as the peaks with the highest frequency among the parietally detected ones.
Statistics
We employed two different statistical tests by using the version 11.1 of the STATISTICA 64 software (StatSoft Inc., Tulsa, USA). First, we tested the relationship of age with the sleep variables, separately in the two groups (WS and TD), by calculating Pearson product moment correlation coefficients between chronological age (years) and sleep architecture/EEG spectra. Next, the dependent variables with significant age-effects were subjected to the homogeneity-of-slopes analysis by using the General Linear Model tool of STATISTICA 64 in order to reveal the possible differences in age-related changes with respect to sleep of WS and TD subjects. The homogeneity-of-slopes model is specifically designed to test whether continuous predictor variables (covariates) have different effects at different levels of categorical independent variables (factors). If the continuous predictor by categorical factor interaction effect is statistically significant, the homogeneity of slopes hypothesis can be rejected. Models included age and the TD/WS variable with the outcome being the sleep indicator. Significant age (continuous predictor in years) group (categorical factor – WS vs. TD) results of such analyses reveal notable differences in aging patterns of sleep in the two groups. In order to clearly and consequently depict the group differences in aging, negative age-effects are visualized by using inverted y-scales (increasing linear trends visualize increased aging). False discovery rate caused by multiple testing (48 Pearson correlations plus 17 interaction effects in the homogeneity of slopes analyses) was controlled by using the Benjamini–Hochberg method. In many applications, including ours this is the desirable control against errors originating from multiplicity (Benjamini & Hochberg, 1995). Consequently, we report the corrected p-values in our paper. 3. Results 3.1. Sleep architecture Sleep architecture was characterized by age-dependent changes in both groups, but the specific variables involved in these effects as well as the extent of the age-effects differed among the groups (Table 1). The correlation coefficients revealing the age-dependent changes in sleep architecture are summarized in Table 1. A significant age-dependent decrease in SWS time and in REM latency was revealed in the TD group (6.05 and 4.36 min/year, respectively). Among the above mentioned age-related changes in the sleep architecture of TD subjects only the age-dependent decrease in SWS time of WS subjects (2.10 min/year) was observed. Apart from the above mentioned age-dependent sleep architectural changes WS subjects were characterized by significant age-related declines in total sleep time (6.8 min/year), sleep efficiency (0.82%/ year), and REM sleep time (2.39 min/year), as well as by age-related increase in WASO (4.03 min/year) and absolute time spent awake during the night (4.86 min/year). In the next step we used homogeneity-of-slopes analyses for the variables with significant age-effects in order to test the differences in the age-related changes in the sleep variables of WS and TD subjects. Significant age group effects were revealed for several sleep architectural variables (Fig. 1) including sleep efficiency (F = 8.61; df = 1, 38; p = .0332; partial
Table 1 Pearson correlation coefficients revealing the age-dependencies of different sleep architectural measures in different groups of subjects. TD Total sleep time (min) Sleep efficiency (%) Wake duration (min) WASO (min) Sleep latency (min) S1 duration (min) S2 duration (min) S3 + S4 (SWS) duration (min) REM duration (min) REM latency (min)

TD – typically developing; WS – Williams syndrome, WASO – wake after sleep onset; S1–S4 – stages of NREM sleep (Stage 1 to Stage 4); SWS – slow wave sleep; REM – rapid eye movement sleep.
* p < .05. ** p < .01. *** p < .001 (Benjamini–Hochberg corrected).
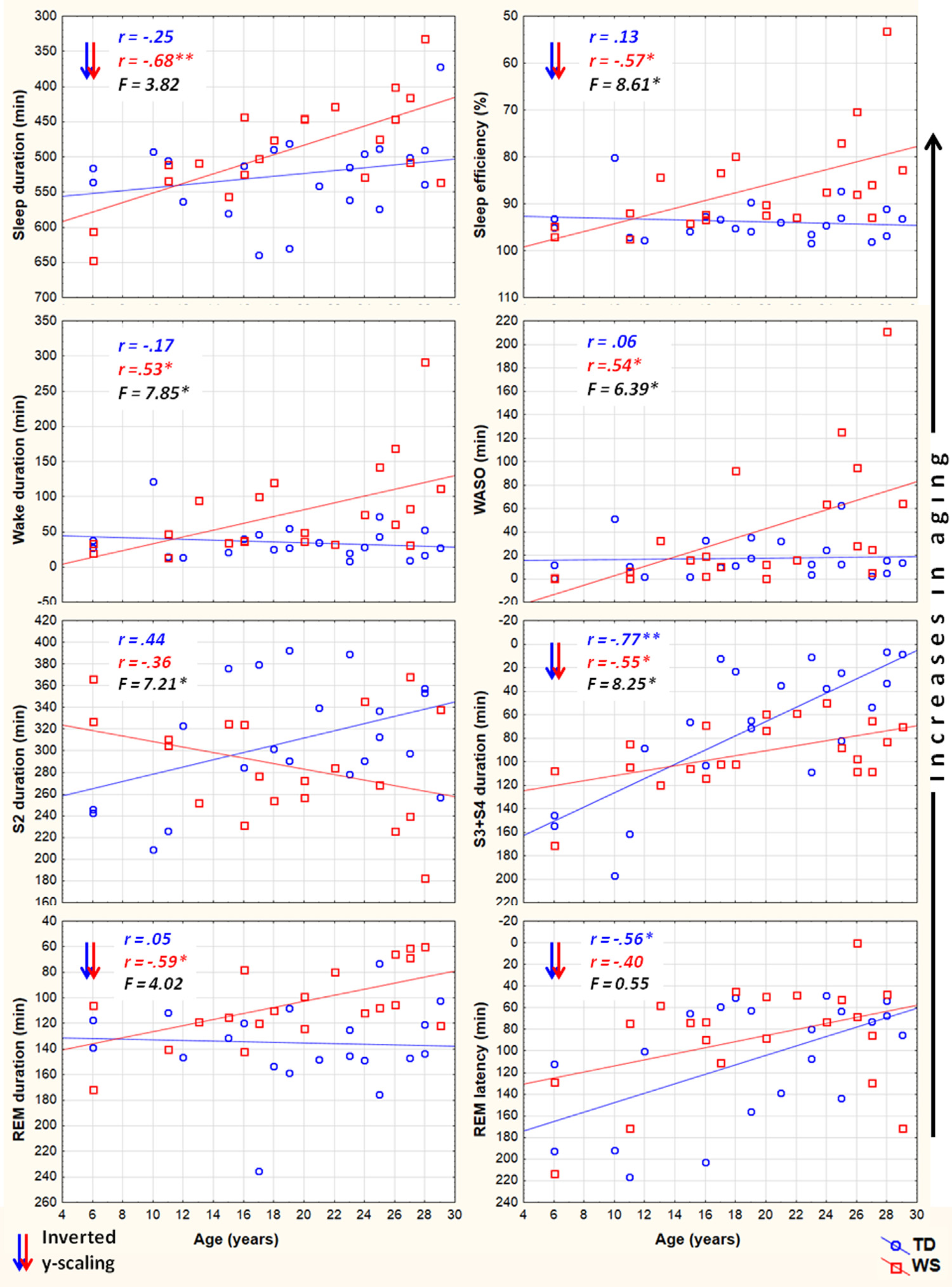
Fig. 1. Aging and sleep architecture in typically developing (TD) and Williams syndrome (WS) subjects. Upward changes depict increasing aging. Inverted scaling for sleep duration, sleep efficiency, S3 + S4 duration, REM duration and REM latency was used. Inserts depict linear relationships as expressed by Pearson correlation coefficients (r), as well as the interaction effects revealed by the homogeneity of slopes analyses (F). *p < .05; **p < .01; ***p < .001.
h2 = .18), wake duration (F = 7.85; df = 1, 38; p = .0368; partial h2 = .17), WASO (F = 6.39; df = 1, 38; p = .0393; partial h2 = .14), S2 duration (F = 7.21; df = 1, 38; p = .0384; partial h2 = .16), and SWS duration (F = 8.25; df = 1, 38; p = .0357; partial h2 = .17). 3.2. Sleep EEG spectra NREM sleep EEG spectra are characterized by age-dependent changes in both TD and WS subjects (Table 2). Delta, theta and alpha band power values are decreasing with age. Significant age-dependent decrease in beta activity was found in TD, but not in WS subjects.
Table 2 Pearson correlation coefficients revealing the age-dependencies of different sleep EEG spectral band power values at derivation Fz.
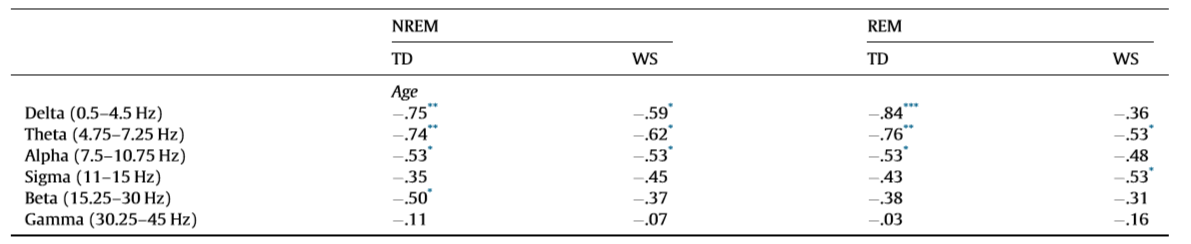
TD – typically developing; WS – Williams syndrome. * p < .05. ** p < .01. *** p < .001 (Benjamini–Hochberg corrected).
Likewise, REM sleep is characterized by age-dependent decreases in EEG delta, theta and alpha power of TD subjects. A slightly dissimilar picture emerged in the WS group, however. REM sleep EEG delta and alpha power of WS subjects was not significantly age-related, but – just as in the case of TD subjects – age correlated negatively with REM sleep theta power. Moreover, a significant negative correlation between WS subjects’ REM sleep EEG sigma power and age was observed (Table 2). Fig. 2 depicts the EEG delta-to-sigma spectral band power values as linear functions of age in TD and in WS subjects. The homogeneity of slopes analysis revealed homogenous age-dependent regression curves for NREM and REM sleep EEG spectral band power values of our groups (WS and TD), as none of the age group interactions proved to be significant (Fig. 2). Likewise, we revealed no age group effect for beta activity in NREM sleep (F = 0.15; df = 1, 38; p = .6919; partial h2 = .004). Group effects on NREM, but not REM sleep EEG spectral band power of an overlapping sample were reported elsewhere (Bódizs et al., 2012), without an explicit depiction of the age effects. 3.3. NREM sleep EEG sigma peak frequencies Among the sigma spectral peak frequencies the fast parietal, but not the slow frontal ones were significantly and linearly related with age in both WS and TD subjects. This significant relationship consisted in an age-dependent increase in the parietally detected fast sigma peak frequencies in TD (r = .61; p = .0252) and WS (r = .55; p = .0363) subjects. There was no significant age group interaction in this measure (F = .00; df = 1, 38; p = .9786; partial h2 = .00). 4. Discussion The tight relationship between age and sleep (Carrier et al., 2001; Ohayon et al., 2004) sheds light on neurodevelopmental/maturational (Colrain & Baker, 2011; Feinberg & Campbell, 2013; Ringli & Huber, 2011; Tarokh et al., 2011) as well as on neurocognitive aging (Espiritu, 2008; Feinberg et al., 1967; Landolt & Borbély, 2001) processes. Many of the known sleep-related indexes of the above mentioned neurodevelopmental and aging effects were tested in the present work. Decreases in the length of night-time sleep amount and changes in its composition are clearly age-dependent (Ohayon et al., 2004) and developmentally relevant (Iglowstein et al., 2003) issues. Moreover, the age-dependent changes in specific spectral content and topography of sleep EEG are reflecting specific maturational (Feinberg & Campbell, 2013) and aging effects (Landolt & Borbély, 2001), related to synaptic pruning or regional differences in brain maturation (Ringli & Huber, 2011). Most of our findings on TD subjects and some of our results on WS subjects were coherent with outcomes of the existing reports. However, specificities and peculiarities of the WS population were also noted. These specificities and peculiarities of age-related changes in polysomnographic and quantitative sleep EEG measures of WS subjects could shed light on the atypical neurodevelopmental processes as well as on the presumed premature aging of subjects characterized by 7q11.23 microdeletion. Results of our current study revealed that a significant premature impairment of sleep structure is an inherent part of WS symptomatology. Several sleep architectural variables (total sleep time, sleep efficiency and REM duration) are characterized by negative age-effects in the WS, but not in the TD group (Table 1) in our current samples ranging from 6 to 29 years of age. The related negative measures of sleep quality (time spent awake and WASO) reveal a complementary picture: agedependent increases were found to be characteristic for WS but not for TD subjects (Table 1) in these polysomnographic measures of sleep impairment. The regression slopes for age vs. sleep efficiency, age vs. wake duration and age vs. WASO are significantly inhomogeneous between the groups (WS and TD, see Fig. 1 and Section 3.1). Age-related sleep impairment in terms of the above measures is well known in the literature of normal human sleep (Ohayon et al., 2004). However, typical timing of the significant changes in these measures of sleep quality is middle age, while our oldest subjects are young adults with a maximum of 29 years of age.

Fig. 2. Aging and sleep electroencephalogram (EEG) power in typically developing (TD) and Williams syndrome (WS) subjects. Upward changes depict increasing aging. Inverted scaling for all variables was used. Inserts depict linear relationships as expressed by Pearson correlation coefficients (r), as well as the interaction effects revealed by the homogeneity of slopes analyses (F). Note the parallelism of the age vs. sleep EEG power functions in the groups, as well as the differences in delta activity (WS > TD). *p < .05; **p < .01; ***p < .001.
The premature aging hypothesis of WS was mainly supported by our findings of an accelerated age-dependent sleep architectural impairment affecting sleep efficiency, wake time and WASO. Possible physiological factors leading to sleep impairment in WS include the reported alterations in cortisol and melatonin release, which were hypothesized to be involved in the pathophysiology and environmental psychophysiology of sleep disturbance in WS (Dimitrio, Sniecinska, & Iles, 2013). Likewise, an age-dependent increase in the prevalence of GAD among subjects with WS might be a cause of premature sleep impairment. Insomnia associated with GAD was shown to be characterized by decreased total sleep time, sleep efficiency, and S2 sleep, as well as by increased SWS time (Saletu et al., 1994). This pattern overlaps significantly with our earlier reports on age-independent (Gombos et al., 2011; Bódizs et al., 2012, 2014) and current findings of age-dependent sleep alterations in WS. Moreover, symptoms of anxiety were shown to be related with alterations in cortisol release patterns (Vreeburg et al., 2010; Hek et al., 2013). Given the facts that cortisol infusion leads to a decrease in REM sleep of human volunteers (Born, Spa¨th-Schwalbe, Schwakenhofer, Kern, & Fehm, 1989) and that unfamiliar settings were shown to increase cortisol release in WS subjects (Lense, Tomarken, & Dykens, 2013), the above indirect evidence is suggestive of an age- and context-dependent increase in the psychosomatic involvement of the hypothalamo–pituitary–adrenal axis in the accentuation of sleep disorders in WS. It should be noted, however, that none of our WS subjects had a diagnosis of GAD, although symptoms of anxiety could still be present in an age-dependent manner. In contrast to the above mentioned sleep efficiency-related measures of sleep quality, NREM sleep-related aspects of aging diverge significantly from the predictions of the premature aging hypothesis of WS. Our results on sleep architecture reveal a decelerated age-dependent decrease in SWS time of WS subjects. The effect is supported by the difference in the magnitudes of the correlations (Table 1), the visual inspection of the scatterplots in the respective field of Fig. 1 and by the significance of the age group interaction effect in the homogeneity-of-slopes analysis. In addition, the above effect is mirrored in our findings on S2 sleep, reflecting the time spent in shallower NREM and non-SWS periods. S2 sleep time is agedependently decreasing in TD, but not in WS subjects (Table 1), while the age group interaction reveals significantly nonhomogenous age vs. S2 sleep regression slopes for the groups (Fig. 1). In line with the above NREM sleep-related macrostructural findings, age-dependent decreases in NREM sleep EEG delta power are not different among the TD and the WS group (Table 2, Fig. 2). Thus, the lower rate of age-dependent SWS decrease in WS subjects is associated with the lack of an age-dependent increase in their S2 duration (Table 1). This peculiar aging effect could reflect an increasing need for a compensation of increasing wakefulness and lost sleep in WS subjects or alternatively a developmental delay in the maturational processes reflected in the increasing shallowness of sleep. Age-dependent decreases in both NREM and REM sleep EEG delta power are evident for the TD group (Table 2). These decreases cohere with previous reports (Feinberg & Campbell, 2013). However, in comparison to the TD group, sleep EEG delta activity of the WS subjects was shown to be increased (Gombos et al., 2011; Bódizs et al., 2012). In technical terms, this continuous (age-independent) increase in sleep EEG delta power results in a clear developmental delay of WS subjects, as the appropriate levels of delta activity are reached during later ages (Fig. 2). Although not entirely parallel with the sleep macrostructural findings, this maturational delay coheres partially with the decelerated age-dependent decrease in the depth of sleep in WS (see above). Taken together, the developmental delays in sleep depth strikingly diverge from the reported findings of an advanced puberty in WS. It seems that advanced puberty is characterized by atypical neurodevelopmental processes in WS as the delays in the age-dependent decrease in sleep depth are not concerted with the hormonal changes inherent to puberty. Origins of this disconcerted developmental/aging pattern of WS subjects could emerge from processes compensating for increasing wakefulness and decreasing sleep efficiency. Possible neurocognitive consequences of this apparent developmental disharmony should be specifically investigated in the near future. Our current findings on significant age-dependencies of several sleep EEG spectral measures co-occurring with similar age-independent displacements of the WS group (Fig. 2) are evident for NREM and REM sleep EEG alpha power, NREM sleep EEG sigma power, as well as the NREM sleep EEG fast parietal sigma spectral peak frequencies (Gombos et al., 2011; Bódizs et al., 2012). Age-dependent decreases in NREM and REM sleep alpha power were found to be significant for both TD and WS subjects (Table 2), but the intercepts of the regression lines as well as the homogeneity of slopes indicate an age-independent deficit in alpha waves of WS subjects (Fig. 2). This is in line with previous findings of a decreased alpha EEG power in WS and needs theoretical explanation (Gombos et al., 2011; Bódizs et al., 2012, 2014). A similar, but less accentuated effect is seen for NREM sleep EEG sigma power (Table 2, Fig. 2). NREM sleep EEG sigma spectral peak frequencies reflect the modal frequency of sleep spindles and were formerly shown to be higher in WS than in TD subjects (Bódizs et al., 2012). Here we show that this difference is continuously present between the groups and is not modulated by chronological age (Section 3.3). Ageindependent alterations in melatonin production, peculiarities in myelination or decreases in basal ganglia volumes could result in the acceleration of the thalamocortical oscillatory dynamics in WS (Bódizs et al., 2012). There are several limitations of our study which has to be mentioned. First of all the number of younger (<14 years of age) subjects is lower than the number of our older age subjects. This is mainly determined by two factors. First, WS is a rare disease and subjects are not easy to found. Moreover, the procedure of all night polysomnography on two consecutive nights is not easily applied to younger children living with developmental disabilities. A lower number of subjects in the lower age range can influence the estimated trends in sleep variables. Thus, further studies conducted on a higher number of subjects are needed in order to increase our knowledge on the peculiarities of age-dependent sleep alterations in WS. Another limitation is the cross-sectional nature of our study. A longitudinal study could be a major step in a more detailed clarification of the atypical aspects of the development and aging of sleep in subjects with WS. Last, but not least, we do not have data on the behavioral phenotype of our subjects. Future studies should include measures of anxiety as this variable is of potential interest in explaining variations in sleep quality. 5. Conclusions In sum, the list of body functions and anatomical signs reflecting the mildly accelerated aging of subjects with WS can be complemented with the premature sleep impairment, characterized by accelerated decreases in sleep efficiency, as well as by early increases in wake time and WASO. Alterations in hypothalamo–pituitary adrenal axis activity, melatonin secretion patterns or other unknown factors could lead to the above effects. In spite of these accelerations in the aging of sleep in subjects with WS, indications for apparent developmental delays in the age-dependent decrease in the depth of sleep were also observed. These findings indicate that early sleep impairment and disharmonic neurodevelopmental processes reflected in sleep structure are significant and clinically relevant aspects of the WS phenotype.
Acknowledgement
This work was supported by the Hungarian National Science Fund (OTKA-NF60806 and OTKA-NK104481 to I.K. and OTKA-PD83876 to J.M.R.).
References
Annaz, D., Hill, C. M., Ashworth, A., Holley, S., & Karmiloff-Smith, A. (2011). Characterisation of sleep problems in children with Williams syndrome. Research in Developmental Disabilities, 32, 164–169.
Arens, R., Wright, B., Elliott, J., Zhao, H., Wang, P. P., Brown, L. W., et al. (1998). Periodic limb movement in sleep in children with Williams syndrome. Journal of Pediatrics, 133, 670–674.
Ashworth, A., Hill, C. M., Karmiloff-Smith, A., & Dimitriou, D. (2013). Cross syndrome comparison of sleep problems in children with Down syndrome and Williams syndrome. Research in Developmental Disabilities, 34, 1572–1580.
Aström, C., & Trojaborg, W. (1992). Relationship of age to power spectrum analysis of EEG during sleep. Journal of Clinical Neurophysiology, 9, 424–430.
Atkinson, J., Anker, S., Braddick, O., Nokes, L., Mason, A., & Braddick, F. (2001). Visual and visuospatial development in young children with Williams syndrome. Developmental Medicine and Child Neurology, 43, 330–337.
Benjamini, Y., & Hochberg, Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B (Methodological), 57, 289–300.
Born, J., Spa¨th-Schwalbe, E., Schwakenhofer, H., Kern, W., & Fehm, H. L. (1989). Influences of corticotropin-releasing hormone, adrenocorticotropin, and cortisol on sleep in normal man. Journal of Clinical Endocrinology & Metabolism, 68, 904–911.
Bódizs, R., Gombos, F., & Kovács, I. (2012). Sleep EEG fingerprints reveal accelerated thalamocortical oscillatory dynamics in Williams syndrome. Research in Developmental Disabilities, 33, 153–164.
Bódizs, R., Gombos, F., Szo˝ cs, K., Réthelyi, J. M., Gerván, P., & Kovács, I. (2014). Sleep-EEG in dizygotic twins discordant for Williams syndrome. Ideggyógyászati Szemle, 67, 59–68.
Carrier, J., Land, S., Buysse, D. J., Kupfer, D. J., & Monk, T. H. (2001). The effects of age and gender on sleep EEG power spectral density in the middle years of life (ages 20–60 years old). Psychophysiology, 38, 232–242.
Cherniske, E. M., Sadler, L. S., Schwartz, D., Carpenter, T. O., & Pober, B. R. (1999). Early puberty in Williams syndrome. Clinical Dysmorphology, 8, 117–121.
Cherniske, E. M., Carpenter, T. O., Klaiman, C., Young, E., Bregman, J., Insogna, K., et al. (2004). Multisystem study of 20 older adults with Williams syndrome. American Journal of Medical Genetetics Part A, 131, 255–264.
Coble, P. A., Kupfer, D. J., Taska, L. S., & Kane, J. (1984). EEG sleep of normal healthy children. Part I: Findings using standard measurement methods. Sleep, 7, 289– 303.
Colrain, I. M., & Baker, F. C. (2011). Changes in sleep as a function of adolescent development. Neuropsychology Review, 21, 5–21.
Crowley, K., Trinder, J., Kim, Y., Carrington, M., & Colrain, I. M. (2002). The effects of normal aging on sleep spindle and K-complex production. Clinical Neurophysiology, 113, 1615–1622.
Darchia, N., Campbell, I. G., Tan, X., & Feinberg, I. (2007). Kinetics of NREM delta EEG power density across NREM periods depend on age and on delta-band designation. Sleep, 30, 71–79.
Devenny, D. A., Krinsky-McHale, S. J., Kittler, P. M., Flory, M., Jenkins, E., & Brown, W. T. (2004). Age-associated memory changes in adults with Williams syndrome. Developmental Neuropsychology, 26, 691–706.
Dimitrio, D., Sniecinska, A., & Iles, R. K. (2013). Abnormal endocrine and behavioural sleep markers in a child with Williams syndrome and siblings. Journal of Sleep Disorders & Therapy, 2, 1000108.
Dykens, E. M. (2003). Anxiety, fears, and phobias in persons with Williams syndrome. Developmental Neuropsychology, 23, 291–316.
Elison, S., Stinton, C., & Howlin, P. (2010). Health and social outcomes in adults with Williams syndrome: Findings from cross-sectional and longitudinal cohorts. Research in Developmental Disabilities, 31, 587–599.
Espiritu, J. R. (2008). Aging-related sleep changes. Clinics in Geriatric Medicine, 24, 1–14. Feinberg, I., Koresko, R. L., & Heller, N. (1967). EEG sleep patterns as a function of normal and pathological aging in man. Journal of Psychiatric Research, 5, 107–144.
Feinberg, I., & Campbell, I. G. (2013). Longitudinal sleep EEG trajectories indicate complex patterns of adolescent brain maturation. American Journal of Physiology – Regulatory, Integrative and Comparative Physiology, 304, R296–R303.
Goldman, S. E., Malow, B. A., Newman, K. D., Roof, E., & Dykens, E. M. (2009). Sleep patterns and daytime sleepiness in adolescents and young adults with Williams syndrome. Journal of Intellectual Disability Research, 53, 182–188.
Gombos, F., Bódizs, R., & Kovács, I. (2011). Atypical sleep architecture and altered EEG spectra in Williams syndrome. Journal of Intellectual Disability Research, 55, 255–262.
Greer, J., Riby, D. M., Hamilitona, C., & Riby, L. M. (2013). Attentional lapse and inhibition control in adults with Williams syndrome. Research in Developmental Disabilities, 34, 4170–4177.
Haas, B. W., & Reiss, A. L. (2012). Social brain development in Williams syndrome: The current status and directions for future research. Frontiers in Psychology, 3, 186.
Hek, K., Direk, N., Newson, R. S., Hofman, A., Hoogendijk, W. J., Mulder, C. L., et al. (2013). Anxiety disorders and salivary cortisol levels in older adults: A population-based study. Psychoneuroendocrinology, 38, 300–305.
Iglowstein, I., Jenni, O. G., Molinari, L., & Largo, R. H. (2003). Sleep duration from infancy to adolescence: Reference values and generational trends. Pediatrics, 111, 302–307.
Jasper, H. H. (1958). Report of the committee on methods of clinical examination in electroencephalography. Electroencephalography and Clinical Neurophysiology, 10, 370–375.
Järvinen-Pasley, A., Bellugi, U., Reilly, J., Mills, D. L., Galaburda, A., Reiss, A. L., et al. (2008). Defining the social phenotype in Williams syndrome: A model for linking gene, the brain, and behavior. Developmental Psychopathology, 20, 1–35.
Krinsky-McHale, S. J., Kittler, P., Brown, W. T., Jenkins, E. C., & Devenny, D. A. (2005). Repetition priming in adults with Williams syndrome: Age-related dissociation between implicit and explicit memory. American Journal of Mental Retardation, 110, 482–496.
Laing, E., Hulme, C., Grant, J., & Karmiloff-Smith, A. (2001). Learning to read in Williams syndrome: Looking beneath the surface of atypical reading development. Journal of Child Psychology and Psychiatry, 42, 729–739.
Landolt, H. P., & Borbély, A. A. (2001). Age-dependent changes in sleep EEG topography. Clinical Neurophysiology, 112, 369–377. Lenoff, H. M., Wang, P., Greenberg, F., & Bellugi, U. (1997). Williams syndrome and the brain. Scientific American, 277, 68–73.
Lense, M. D., Tomarken, A. J., & Dykens, E. M. (2013). Diurnal cortisol profile in Williams syndrome in novel and familiar settings. American Journal of Intellectual and Developmental Disabilities, 118, 201–210.
Leyfer, O. T., Woodruff-Borden, J., Klein-Tasman, B. P., Fricke, J. S., & Mervis, C. B. (2006). Prevalence of psychiatric disorders in 4 to 16-year-olds with Williams syndrome. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 141B, 615–622.
Mari, A., Amati, F., Mingarelli, R., Giannotti, A., Sebastio, G., Colloridi, V., et al. (1995). Analysis of the elastin gene in 60 patients with clinical diagnosis of Williams syndrome. Human Genetics, 96, 444–448.
Marler, J. A., Elfenbein, J. L., Ryals, B. M., Urban, Z., & Netzloff, M. L. (2005). Sensorineural hearing loss in children and adults with Williams syndrome. American Journal of Medical Genetics Part A, 138, 318–327.
Mason, T. B., Arens, R., Sharman, J., Bintliff-Janisak, B., Schultz, B., Walters, A. S., et al. (2011). Sleep in children with Williams syndrome. Sleep Medicine, 12, 892– 897.
Meyer-Lindenberg, A., Mervis, C. B., & Berman, K. F. (2006). Neural mechanisms in Williams syndrome: A unique window to genetic influences on cognition and behaviour. Nature Reviews Neuroscience, 7, 380–393.
Morris, C. A., Demsey, S. A., Leonard, C. O., Dilts, C., & Blackburn, B. L. (1988). Natural history of Williams syndrome: Physical characteristics. Journal of Pediatrics, 113, 318–326.
Nicolas, A., Petit, D., Rompré, S., & Montplaisir, J. (2001). Sleep spindle characteristics in healthy subjects of different age groups. Clinical Neurophysiology, 112, 521– 527.
Ohayon, M. M., Carskadon, M. A., Guilleminault, C., & Vitiello, M. V. (2004). Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: Developing normative sleep values across the human lifespan. Sleep, 27, 1255–1273.
Partsch, C. J., Dreyer, G., Gosch, A., Winter, M., Schneppenheim, R., Wessel, A., et al. (1999). Longitudinal evaluation of growth, puberty, and bone maturation in children with Williams syndrome. Journal of Pediatrics, 134, 82–89.
Pober, B. R. (2010). Williams–Beuren syndrome. New England Journal of Medicine, 362, 239–252.
Principe, J. C., & Smith, J. R. (1982). Sleep spindle characteristics as a function of age. Sleep, 5, 73–84.
Rechtschaffen, A., & Kales, A. (1968). Manual of standardized terminology, techniques and scoring system for sleep stages of human participants. Los Angeles: UCLA Brain Information Service/Brain Research Institute.
Ringli, M., & Huber, R. (2011). Developmental aspects of sleep slow waves: Linking sleep, brain maturation and behavior. Progress in Brain Research, 193, 63–82.
Saletu, B., Anderer, P., Brandstätter, N., Frey, R., Grünberger, J., Klösch, G., et al. (1994). Insomnia in generalized anxiety disorder: Polysomnographic, psychometric and clinical investigations before, during and after therapy with a long- versus a short-half-life benzodiazepine (quazepam versus triazolam). Neuropsychobiology, 29, 69–90.
Tarokh, L., Carskadon, M. A., & Achermann, P. (2011). Trait-like characteristics of the sleep EEG across adolescent development. Journal of Neuroscience, 31, 6371– 6378.
Tsai, S. W., Wu, S. K., Liou, Y. M., & Shu, S. G. (2008). Early development in Williams syndrome. Pediatrics International, 50, 221–224.
Vreeburg, S. A., Zitman, F. G., van Pelt, J., Derijk, R. H., Verhagen, J. C., van Dyck, R., et al. (2010). Salivary cortisol levels in persons with and without different anxiety disorders. Psychosomatic Medicine, 72, 340–347.
Wan, E. S., Pober, B. R., Washko, G. R., Raby, B. A., & Silverman, E. K. (2010). Pulmonary function and emphysema in Williams–Beuren syndrome. American Journal of Medical Genetics Part A, 152A, 653–656.